Ιατρικός εμπειρογνώμονας του άρθρου

Νέες δημοσιεύσεις
Νόσος Charcot-Marie-Tooth.
Τελευταία επισκόπηση: 12.07.2025

Όλα τα περιεχόμενα του iLive ελέγχονται ιατρικά ή ελέγχονται για να διασφαλιστεί η όσο το δυνατόν ακριβέστερη ακρίβεια.
Έχουμε αυστηρές κατευθυντήριες γραμμές προμήθειας και συνδέουμε μόνο με αξιόπιστους δικτυακούς τόπους πολυμέσων, ακαδημαϊκά ερευνητικά ιδρύματα και, όπου είναι δυνατόν, ιατρικά επισκοπικά μελέτες. Σημειώστε ότι οι αριθμοί στις παρενθέσεις ([1], [2], κλπ.) Είναι σύνδεσμοι με τις οποίες μπορείτε να κάνετε κλικ σε αυτές τις μελέτες.
Εάν πιστεύετε ότι κάποιο από το περιεχόμενό μας είναι ανακριβές, παρωχημένο ή αμφισβητήσιμο, παρακαλώ επιλέξτε το και πατήστε Ctrl + Enter.
Η περονιαία μυϊκή ατροφία, σύνδρομο ή νόσος Charcot-Marie-Tooth, είναι μια ομάδα χρόνιων κληρονομικών ασθενειών με βλάβη στα περιφερικά νεύρα.
Σύμφωνα με το ICD-10, στην ενότητα για τις ασθένειες του νευρικού συστήματος, ο κωδικός για αυτήν την ασθένεια είναι G60.0 (κληρονομική κινητική και αισθητηριακή νευροπάθεια). Περιλαμβάνεται επίσης στον κατάλογο των ορφανών ασθενειών.
Επιδημιολογία
Σύμφωνα με κλινικές στατιστικές, η επικράτηση όλων των τύπων νόσου Charcot-Marie-Tooth ανά 100 χιλιάδες πληθυσμού είναι 19 περιπτώσεις (σύμφωνα με άλλες πηγές, μία περίπτωση ανά 2,5-10 χιλιάδες πληθυσμού).
Η CMT τύπου 1 ευθύνεται για περίπου τα δύο τρίτα των περιπτώσεων (μία περίπτωση ανά 5.000 έως 7.000 κατοίκους) και σχεδόν το 70% αυτών σχετίζεται με διπλασιασμό του γονιδίου PMP22. Περισσότεροι από 1,2 εκατομμύρια άνθρωποι παγκοσμίως πάσχουν από αυτόν τον τύπο νόσου.
Η συχνότητα εμφάνισης της CMT τύπου 4 εκτιμάται σε 1-5 περιπτώσεις ανά 10.000 παιδιά. [ 1 ]
Αιτίες Νόσος Charcot-Marie-Tooth
Σύμφωνα με την ταξινόμηση των πολυνευροπαθητικών συνδρόμων, η περονιαία (περονιαία) μυϊκή ατροφία, η νευρική αμυοτροφία Charcot-Marie-Tooth ή η νόσος Charcot-Marie-Tooth (συντομογραφία CMT) αναφέρεται σε γενετικά καθορισμένες κινητικές-αισθητηριακές πολυνευροπάθειες. [ 2 ]
Δηλαδή, οι αιτίες εμφάνισής του είναι οι γενετικές μεταλλάξεις. Και ανάλογα με τη φύση των γενετικών αποκλίσεων, διακρίνονται οι κύριοι τύποι ή είδη αυτού του συνδρόμου: απομυελινωτικές και αξονικές. Η πρώτη ομάδα περιλαμβάνει τη νόσο Charcot-Marie-Tooth τύπου 1 (CMT1), η οποία εμφανίζεται λόγω διπλασιασμού του γονιδίου PMP22 στο χρωμόσωμα 17, το οποίο κωδικοποιεί την διαμεμβρανική περιφερική πρωτεΐνη μυελίνης 22. Ως αποτέλεσμα, εμφανίζεται τμηματική απομυελίνωση του ελύτρου του νευρικού συστήματος (αποφύσεις των νευρικών κυττάρων) και μείωση της ταχύτητας αγωγιμότητας του νευρικού σήματος. Επιπλέον, μεταλλάξεις μπορεί να υπάρχουν σε ορισμένα άλλα γονίδια.
Η αξονική μορφή είναι η νόσος Charcot-Marie-Tooth τύπου 2 (CMT2), η οποία επηρεάζει τους ίδιους τους νευράξονες και σχετίζεται με παθολογικές αλλαγές στο γονίδιο MFN2 στον τόπο 1p36.22, που κωδικοποιεί την πρωτεΐνη μεμβράνης μιτοφουσίνη-2, η οποία είναι απαραίτητη για τη σύντηξη των μιτοχονδρίων και τον σχηματισμό λειτουργικών μιτοχονδριακών δικτύων μέσα στα περιφερικά νευρικά κύτταρα. Υπάρχουν περισσότεροι από δώδεκα υποτύποι CMT2 (με μεταλλάξεις σε συγκεκριμένα γονίδια).
Πρέπει να σημειωθεί ότι επί του παρόντος έχουν εντοπιστεί περισσότερα από εκατό γονίδια των οποίων η βλάβη, που μεταδίδεται κληρονομικά, προκαλεί διάφορους υποτύπους της νόσου Charcot-Marie-Tooth. Για παράδειγμα, μεταλλάξεις στο γονίδιο RAB7 οδηγούν σε CMT τύπου 2Β. η αλλοίωση του γονιδίου SH3TC2 (που κωδικοποιεί μία από τις πρωτεΐνες μεμβράνης των κυττάρων Schwann) προκαλεί CMT τύπου 4C, η οποία εκδηλώνεται στην παιδική ηλικία και χαρακτηρίζεται από απομυελίνωση κινητικών και αισθητήριων νευρώνων (υπάρχουν δώδεκα και μισή μορφές τύπου 4 αυτής της νόσου).
Μια σπάνια CMT τύπου 3 (που ονομάζεται σύνδρομο Dejerine-Sottas) αρχίζει να αναπτύσσεται στην πρώιμη παιδική ηλικία και προκαλείται από μεταλλάξεις στα γονίδια PMP22, MPZ, EGR2 και άλλα.
Όταν η CMT τύπου 5 εμφανίζεται στην ηλικία των 5-12 ετών, παρατηρείται όχι μόνο κινητική νευροπάθεια (με τη μορφή σπαστικής παραπάρεσης των κάτω άκρων), αλλά και βλάβη στα οπτικά και ακουστικά νεύρα.
Η μυϊκή αδυναμία και η οπτική ατροφία (με απώλεια όρασης), καθώς και τα προβλήματα ισορροπίας είναι τυπικά της CMT τύπου 6. Και στη νόσο Charcot-Marie-Tooth τύπου 7 δεν υπάρχει μόνο κινητική-αισθητηριακή νευροπάθεια, αλλά και μια νόσος του αμφιβληστροειδούς με τη μορφή μελαγχρωστικής αμφιβληστροειδοπάθειας.
Πιο συχνή στους άνδρες, η φυλοσύνδετη CMT ή νόσος Charcot-Marie-Tooth με τετραπάρεση των άκρων (αδυναμία κίνησης και των δύο χεριών και των ποδιών) είναι ένας απομυελινωτικός τύπος και πιστεύεται ότι προκύπτει από μια μετάλλαξη στο γονίδιο GJB1 στον μακρύ βραχίονα του χρωμοσώματος Χ, το οποίο κωδικοποιεί τη κοννεξίνη 32, μια διαμεμβρανική πρωτεΐνη των κυττάρων Schwann και των ολιγοδενδροκυττάρων που ρυθμίζει τη μετάδοση των νευρικών σημάτων. [ 3 ]
Παράγοντες κινδύνου
Ο κύριος παράγοντας κινδύνου για την CMT είναι το οικογενειακό ιστορικό της νόσου, δηλαδή σε στενούς συγγενείς.
Σύμφωνα με τους γενετιστές, εάν και οι δύο γονείς είναι φορείς του αυτοσωμικού υπολειπόμενου γονιδίου για τη νόσο Charcot-Marie-Tooth, ο κίνδυνος να αποκτήσει ένα παιδί που θα αναπτύξει αυτή τη νόσο είναι 25%. Και ο κίνδυνος το παιδί να είναι φορέας αυτού του γονιδίου (αλλά να μην έχει συμπτώματα) εκτιμάται σε 50%.
Στην περίπτωση της φυλοσύνδετης κληρονομικότητας (όταν το μεταλλαγμένο γονίδιο βρίσκεται στο χρωμόσωμα Χ της γυναίκας), υπάρχει 50% πιθανότητα η μητέρα να μεταβιβάσει το γονίδιο στον γιο της, ο οποίος θα αναπτύξει CMT. Η νόσος μπορεί να μην εμφανιστεί όταν γεννηθεί ένα κορίτσι, αλλά οι γιοι (εγγόνια) της κόρης μπορεί να κληρονομήσουν το ελαττωματικό γονίδιο και να αναπτυχθεί η νόσος.
Παθογένεση
Σε κάθε τύπο νόσου Charcot-Marie-Tooth, η παθογένεσή της προκαλείται από μια κληρονομική ανωμαλία των περιφερικών νεύρων: κινητικά (κινητικά) και αισθητικά (ευαίσθητα).
Εάν ο τύπος CMT είναι απομυελινωτικός, τότε η καταστροφή ή το ελάττωμα του ελύτρου μυελίνης που προστατεύει τους άξονες των περιφερικών νεύρων οδηγεί σε επιβράδυνση της μετάδοσης των νευρικών ερεθισμάτων στο περιφερικό νευρικό σύστημα - μεταξύ του εγκεφάλου, των μυών και των αισθητηρίων οργάνων.
Στον αξονικό τύπο της νόσου, επηρεάζονται οι ίδιοι οι άξονες, γεγονός που επηρεάζει αρνητικά την ισχύ των νευρικών σημάτων, η οποία δεν επαρκεί για την πλήρη διέγερση των μυών και των αισθητηρίων οργάνων.
Διαβάστε επίσης:
Πώς μεταδίδεται το σύνδρομο Charcot-Marie-Tooth; Τα ελαττωματικά γονίδια μπορούν να κληρονομηθούν με αυτοσωματικό επικρατή ή αυτοσωματικό υπολειπόμενο τρόπο.
Ο πιο συνηθισμένος τύπος κληρονομικότητας, η αυτοσωμική επικρατής κληρονομικότητα, συμβαίνει όταν υπάρχει ένα αντίγραφο του μεταλλαγμένου γονιδίου (που φέρει ο ένας από τους δύο γονείς). Και η πιθανότητα μετάδοσης της CMT σε κάθε παιδί που γεννιέται εκτιμάται στο 50%. [ 4 ]
Με την αυτοσωμική υπολειπόμενη κληρονομικότητα, χρειάζονται δύο αντίγραφα του ελαττωματικού γονιδίου (ένα από κάθε γονέα που δεν εμφανίζει κανένα σημάδι της νόσου) για να αναπτυχθεί η νόσος.
Σε 40-50% των περιπτώσεων, εμφανίζεται αυτοσωμική επικρατής κληρονομική απομυελίνωση, δηλαδή CMT τύπου 1. σε 12-26% των περιπτώσεων, αξονική CMT, δηλαδή τύπου 2. Και σε 10-15% των περιπτώσεων παρατηρείται φυλοσύνδετη κληρονομικότητα. [ 5 ]
Συμπτώματα Νόσος Charcot-Marie-Tooth
Συνήθως, τα πρώτα σημάδια αυτής της νόσου αρχίζουν να εμφανίζονται στην παιδική ηλικία και την εφηβεία και αναπτύσσονται σταδιακά καθ' όλη τη διάρκεια της ζωής, αν και το σύνδρομο μπορεί να γίνει αισθητό αργότερα. Ο συνδυασμός των συμπτωμάτων είναι μεταβλητός και ο ρυθμός εξέλιξης της νόσου, καθώς και η σοβαρότητά της, είναι αδύνατο να προβλεφθούν.
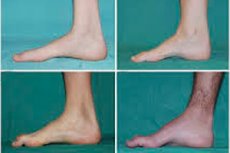
Τυπικά συμπτώματα του αρχικού σταδίου περιλαμβάνουν αυξημένη γενική κόπωση, μειωμένο τόνο (αδυναμία) των μυών των ποδιών, των αστραγάλων και των κνημών, έλλειψη αντανακλαστικών. Αυτό δυσχεραίνει την κίνηση των ποδιών και οδηγεί σε δυσβασία (διαταραχή βάδισης) με τη μορφή υψηλότερης ανύψωσης των ποδιών, συχνά με συχνά σκοντάφτισμα και πτώσεις. Τα σημάδια της νόσου Charcot-Marie-Tooth σε ένα μικρό παιδί μπορεί να περιλαμβάνουν έντονη αδεξιότητα και ασυνήθιστες για την ηλικία δυσκολίες στο περπάτημα που σχετίζονται με αμφοτερόπλευρη πτώση του ποδιού. Χαρακτηριστικές είναι επίσης οι παραμορφώσεις των ποδιών: υψηλή καμάρα (κούφιο πόδι) ή σοβαρή πλατυποδία, κυρτά (σχήματος σφυριού) δάχτυλα.
Στην περίπτωση του περπατήματος στα δάχτυλα των ποδιών με φόντο μυϊκή υποτονία, ένας νευρολόγος μπορεί να υποψιάζεται ότι το παιδί έχει CMT τύπου 4, στην οποία τα παιδιά μπορεί να μην μπορούν να περπατήσουν μέχρι την εφηβεία.
Καθώς η νόσος εξελίσσεται, η μυϊκή ατροφία και η αδυναμία εξαπλώνονται στα άνω άκρα, καθιστώντας δύσκολη την εκτέλεση λεπτών κινητικών δεξιοτήτων και την εκτέλεση κανονικών εργασιών με τα χέρια. Η μειωμένη αίσθηση αφής και η ικανότητα να αισθάνεται κανείς τη θερμότητα και το κρύο, καθώς και το μούδιασμα στα πόδια και τα χέρια, υποδηλώνουν βλάβη στους άξονες των αισθητήριων νεύρων.
Στη νόσο Charcot-Marie-Tooth τύπου 3 και 6 που εκδηλώνεται στην παιδική ηλικία, παρατηρούνται αισθητική αταξία (διαταραχή του συντονισμού των κινήσεων και της ισορροπίας), μυϊκές συσπάσεις και τρόμος, βλάβη στο προσωπικό νεύρο, ατροφία του οπτικού νεύρου με νυσταγμό και απώλεια ακοής.
Σε μεταγενέστερα στάδια, μπορεί να υπάρχει ανεξέλεγκτο τρέμουλο (τρόμος) και συχνές μυϊκές κράμπες. Τα προβλήματα με την κίνηση μπορούν να οδηγήσουν στην ανάπτυξη πόνου: μυϊκού, αρθρικού, νευροπαθητικού.
Επιπλοκές και συνέπειες
Η νόσος Charcot-Marie-Tooth μπορεί να έχει επιπλοκές και συνέπειες όπως:
- συχνότερα διαστρέμματα και κατάγματα.
- συσπάσεις που σχετίζονται με τη βράχυνση των περιαρθρικών μυών και τενόντων.
- σκολίωση (καμπυλότητα της σπονδυλικής στήλης);
- αναπνευστικά προβλήματα – όταν οι νευρικές ίνες που νευρώνουν τους μύες του διαφράγματος έχουν υποστεί βλάβη:
- απώλεια της ικανότητας ανεξάρτητης κίνησης.
Διαγνωστικά Νόσος Charcot-Marie-Tooth
Η διάγνωση περιλαμβάνει κλινική εξέταση, αναμνησία (συμπεριλαμβανομένου του οικογενειακού ιστορικού), νευρολογική και συστηματική εξέταση.
Διεξάγονται εξετάσεις για τον έλεγχο του εύρους κίνησης, της ευαισθησίας και των αντανακλαστικών των τενόντων. Η νευρική αγωγιμότητα μπορεί να αξιολογηθεί χρησιμοποιώντας διαγνωστικά όργανα – ηλεκτρομυογράφημα ή ηλεκτρονευρομυογράφημα. Μπορεί επίσης να απαιτηθεί υπερηχογράφημα ή μαγνητική τομογραφία. [ 6 ]
Οι γενετικές εξετάσεις ή οι εξετάσεις DNA για την ανίχνευση των πιο συνηθισμένων γενετικών μεταλλάξεων που προκαλούν CMT σε ένα δείγμα αίματος είναι περιορισμένες, επειδή οι εξετάσεις DNA δεν είναι προς το παρόν διαθέσιμες για όλους τους τύπους της νόσου. Για περισσότερες πληροφορίες, ανατρέξτε στην ενότητα Γενετικές εξετάσεις.
Σε ορισμένες περιπτώσεις, πραγματοποιείται βιοψία του περιφερικού νεύρου (συνήθως του συρικού νεύρου).
Διαφορική διάγνωση
Η διαφορική διάγνωση περιλαμβάνει άλλες περιφερικές νευροπάθειες, μυϊκή δυστροφία Duchenne, μυελοπαθητικά και μυασθενικά σύνδρομα, διαβητική νευροπάθεια, μυελοπάθειες στην πολλαπλή και αμυοτροφική πλευρική σκλήρυνση, σύνδρομο Guillain-Barré, τραύμα στο περονιαίο νεύρο και την ατροφία του (συμπεριλαμβανομένης της σύσπασης μεταξύ των οσφυϊκών δίσκων της σπονδυλικής στήλης), βλάβη στην παρεγκεφαλίδα ή τον θάλαμο, καθώς και παρενέργειες της χημειοθεραπείας (κατά τη διάρκεια της θεραπείας με κυτταροστατικά όπως η βινκριστίνη ή η πακλιταξέλη). [ 7 ]
Ποιος θα επικοινωνήσει;
Θεραπεία Νόσος Charcot-Marie-Tooth
Σήμερα, η θεραπεία για αυτή την κληρονομική ασθένεια περιλαμβάνει ασκησιοθεραπεία (που στοχεύει στην ενδυνάμωση και το τέντωμα των μυών), εργοθεραπεία (η οποία βοηθά τους ασθενείς με μυϊκή αδυναμία στα χέρια) και τη χρήση ορθοπεδικών βοηθημάτων για τη διευκόλυνση του βαδίσματος. Εάν είναι απαραίτητο, συνταγογραφούνται παυσίπονα ή αντισπασμωδικά. [ 8 ]
Σε περιπτώσεις σοβαρής πλατυποδίας, μπορεί να πραγματοποιηθεί οστεοτομή, και σε περίπτωση παραμόρφωσης της πτέρνας, ενδείκνυται χειρουργική διόρθωση – αρθρόδεση. [ 9 ]
Η έρευνα βρίσκεται σε εξέλιξη τόσο για τη γενετική συνιστώσα της νόσου όσο και για τις μεθόδους θεραπείας της. Η χρήση βλαστοκυττάρων, ορισμένων ορμονών, λεκιθίνης ή ασκορβικού οξέος δεν έχει ακόμη αποφέρει θετικά αποτελέσματα.
Χάρη όμως σε πρόσφατη έρευνα, κάτι νέο μπορεί πράγματι να εμφανιστεί στη θεραπεία της νόσου Charcot-Marie-Tooth στο εγγύς μέλλον. Έτσι, από το 2014, η γαλλική εταιρεία Pharnext αναπτύσσει και από τα μέσα του 2019 βρίσκονται σε εξέλιξη κλινικές δοκιμές για το φάρμακο PXT3003 για τη θεραπεία της CMT τύπου 1 σε ενήλικες, το οποίο καταστέλλει την αυξημένη έκφραση του γονιδίου PMP22, βελτιώνει τη μυελίνωση των περιφερικών νεύρων και ανακουφίζει από τα νευρομυϊκά συμπτώματα.
Ειδικοί από την ιατρική εταιρεία Sarepta Therapeutics (ΗΠΑ) εργάζονται για τη δημιουργία μιας γονιδιακής θεραπείας για τη νόσο Charcot-Marie-Tooth τύπου 1. Αυτή η θεραπεία θα χρησιμοποιήσει έναν ακίνδυνο αδενο-σχετιζόμενο ιό (AAV) του γένους Dependovirus με γραμμικό μονοκλωνικό γονιδίωμα DNA, το οποίο θα μεταφέρει το γονίδιο NTF3 στον οργανισμό, κωδικοποιώντας την πρωτεΐνη νευροτροφίνη-3 (NT-3) που είναι απαραίτητη για τη λειτουργία των νευρικών κυττάρων Schwann.
Η Helixmith θα ξεκινήσει κλινικές δοκιμές της γονιδιακής θεραπείας Engensis (VM202) που αναπτύχθηκε στη Νότια Κορέα μέχρι το τέλος του 2020 για τη θεραπεία μυϊκών συμπτωμάτων στη CMT τύπου 1. [ 10 ]
Πρόληψη
Η πρόληψη της CMT μπορεί να γίνει με τη γενετική συμβουλευτική των μελλοντικών γονέων, ειδικά εάν κάποιος στο ζευγάρι έχει οικογενειακό ιστορικό της νόσου. Ωστόσο, έχουν εντοπιστεί περιπτώσεις μεταλλάξεων de novo σημειακών γονιδίων, δηλαδή, απουσία της νόσου στο οικογενειακό ιστορικό.
Κατά τη διάρκεια της εγκυμοσύνης, η πιθανότητα εμφάνισης νόσου Charcot-Marie-Tooth στο μελλοντικό παιδί μπορεί να ελεγχθεί με βιοψία χοριακών λάχνων (από 10 έως 13 εβδομάδες κύησης), καθώς και με ανάλυση αμνιακού υγρού (στις 15-18 εβδομάδες).
Πρόβλεψη
Γενικά, η πρόγνωση για διαφορετικούς τύπους νόσου Charcot-Marie-Tooth εξαρτάται από τη σοβαρότητα της κλινικής εικόνας, αλλά σε όλες τις περιπτώσεις η νόσος εξελίσσεται αργά. Πολλοί ασθενείς έχουν αναπηρίες, αν και αυτό δεν μειώνει το προσδόκιμο ζωής.