Ιατρικός εμπειρογνώμονας του άρθρου

Νέες δημοσιεύσεις
Κληρονομική νεφρίτιδα (σύνδρομο Alport) στα παιδιά
Τελευταία επισκόπηση: 05.07.2025

Όλα τα περιεχόμενα του iLive ελέγχονται ιατρικά ή ελέγχονται για να διασφαλιστεί η όσο το δυνατόν ακριβέστερη ακρίβεια.
Έχουμε αυστηρές κατευθυντήριες γραμμές προμήθειας και συνδέουμε μόνο με αξιόπιστους δικτυακούς τόπους πολυμέσων, ακαδημαϊκά ερευνητικά ιδρύματα και, όπου είναι δυνατόν, ιατρικά επισκοπικά μελέτες. Σημειώστε ότι οι αριθμοί στις παρενθέσεις ([1], [2], κλπ.) Είναι σύνδεσμοι με τις οποίες μπορείτε να κάνετε κλικ σε αυτές τις μελέτες.
Εάν πιστεύετε ότι κάποιο από το περιεχόμενό μας είναι ανακριβές, παρωχημένο ή αμφισβητήσιμο, παρακαλώ επιλέξτε το και πατήστε Ctrl + Enter.
Η κληρονομική νεφρίτιδα (σύνδρομο Alport) είναι μια γενετικά καθορισμένη κληρονομική μη ανοσολογική σπειραματοπάθεια, που εκδηλώνεται με αιματουρία (μερικές φορές με πρωτεϊνουρία), προοδευτική μείωση της νεφρικής λειτουργίας με την ανάπτυξη χρόνιας νεφρικής ανεπάρκειας, συχνά σε συνδυασμό με αισθητηριακή κώφωση και οπτική βλάβη.
Η ασθένεια περιγράφηκε για πρώτη φορά το 1902 από τον LG Guthrie, ο οποίος παρατήρησε μια οικογένεια στην οποία παρατηρήθηκε αιματουρία σε αρκετές γενιές. Το 1915, ο AF Hurst περιέγραψε την ανάπτυξη ουραιμίας σε μέλη της ίδιας οικογένειας. Το 1927, ο A. Alport εντόπισε για πρώτη φορά απώλεια ακοής σε αρκετούς συγγενείς με αιματουρία. Τη δεκαετία του 1950, περιγράφηκαν οφθαλμικές αλλοιώσεις σε παρόμοια ασθένεια. Το 1972, σε ασθενείς με κληρονομική αιματουρία, κατά τη διάρκεια μιας μορφολογικής μελέτης νεφρικού ιστού, οι Hinglais et al. αποκάλυψαν ανομοιόμορφη επέκταση και διαστρωμάτωση των σπειραματικών βασικών μεμβρανών. Το 1985, εντοπίστηκε η γενετική βάση της κληρονομικής νεφρίτιδας - μια μετάλλαξη στο γονίδιο του κολλαγόνου τύπου IV (Fiengold et al., 1985).
Η μελέτη της γενετικής φύσης της νόσου μας επέτρεψε να συμπεράνουμε ότι οι διαφορές στις φαινοτυπικές εκδηλώσεις της κληρονομικής νεφρίτιδας (με ή χωρίς απώλεια ακοής) οφείλονται στον βαθμό έκφρασης του μεταλλαγμένου γονιδίου. Έτσι, προς το παρόν, όλες οι κλινικές παραλλαγές θεωρούνται ως εκδηλώσεις μίας νόσου και ο όρος «κληρονομική νεφρίτιδα» είναι συνώνυμος με τον όρο «σύνδρομο Alport».
Σύμφωνα με επιδημιολογικές μελέτες, η κληρονομική νεφρίτιδα εμφανίζεται με συχνότητα 17 ανά 100.000 παιδιά.
 [
[ Αιτίες του συνδρόμου Alport
Η γενετική βάση της νόσου είναι μια μετάλλαξη στο γονίδιο της αλυσίδας α-5 του κολλαγόνου τύπου IV. Αυτός ο τύπος είναι καθολικός για τις βασικές μεμβράνες του νεφρού, την κοχλιακή συσκευή, την κάψουλα του φακού, τον αμφιβληστροειδή και τον κερατοειδή του οφθαλμού, κάτι που έχει αποδειχθεί σε μελέτες που χρησιμοποιούν μονοκλωνικά αντισώματα έναντι αυτού του κλάσματος κολλαγόνου. Πρόσφατα, έχει υποδειχθεί η δυνατότητα χρήσης ανιχνευτών DNA για την προγεννητική διάγνωση της κληρονομικής νεφρίτιδας.
Τονίζεται η σημασία της εξέτασης όλων των μελών της οικογένειας με ανιχνευτές DNA για την αναγνώριση φορέων του μεταλλαγμένου γονιδίου, κάτι που έχει μεγάλη σημασία στη διεξαγωγή ιατρικής και γενετικής συμβουλευτικής σε οικογένειες με αυτή την ασθένεια. Ωστόσο, έως και 20% των οικογενειών δεν έχουν συγγενείς που πάσχουν από νεφρική νόσο, γεγονός που υποδηλώνει υψηλή συχνότητα αυθόρμητων μεταλλάξεων του μη φυσιολογικού γονιδίου. Οι περισσότεροι ασθενείς με κληρονομική νεφρίτιδα έχουν άτομα με νεφρική νόσο, απώλεια ακοής και παθολογία όρασης στις οικογένειές τους. Οι γάμοι εξ αίματος μεταξύ ατόμων με έναν ή περισσότερους προγόνους είναι σημαντικοί, καθώς στον γάμο συγγενών ατόμων αυξάνεται η πιθανότητα λήψης των ίδιων γονιδίων και από τους δύο γονείς. Έχουν καθιερωθεί αυτοσωμικές κυρίαρχες, αυτοσωμικές υπολειπόμενες και κυρίαρχες, φυλοσύνδετες οδοί μετάδοσης.
Στα παιδιά, διακρίνονται συνήθως τρεις τύποι κληρονομικής νεφρίτιδας: το σύνδρομο Alport, η κληρονομική νεφρίτιδα χωρίς απώλεια ακοής και η οικογενής καλοήθης αιματουρία.
Το σύνδρομο Alport είναι μια κληρονομική νεφρίτιδα με εξασθένηση της ακοής. Βασίζεται σε ένα συνδυασμένο ελάττωμα στη δομή του κολλαγόνου της σπειραματικής βασικής μεμβράνης των νεφρών, των αυτιών και των οφθαλμικών δομών. Το γονίδιο του κλασικού συνδρόμου Alport βρίσκεται στον τόπο 21-22 q του μακρού βραχίονα του χρωμοσώματος Χ. Στις περισσότερες περιπτώσεις, κληρονομείται με κυρίαρχο τρόπο, συνδεδεμένο με το χρωμόσωμα Χ. Από αυτή την άποψη, το σύνδρομο Alport είναι πιο σοβαρό στους άνδρες, καθώς στις γυναίκες η λειτουργία του μεταλλαγμένου γονιδίου αντισταθμίζεται από ένα υγιές αλληλόμορφο του δεύτερου, άθικτου χρωμοσώματος.
Η γενετική βάση για την ανάπτυξη της κληρονομικής νεφρίτιδας είναι μεταλλάξεις στα γονίδια των αλυσίδων άλφα του κολλαγόνου τύπου IV. Είναι γνωστές έξι αλυσίδες άλφα του κολλαγόνου τύπου IV G: τα γονίδια των αλυσίδων α5 και α6 (Col4A5 και Col4A5) βρίσκονται στον μακρύ βραχίονα του χρωμοσώματος Χ στη ζώνη 21-22q. τα γονίδια των αλυσίδων α3 και α4 (Col4A3 και Col4A4) βρίσκονται στο 2ο χρωμόσωμα. τα γονίδια των αλυσίδων α1 και α2 (Col4A1 και Col4A2) βρίσκονται στο 13ο χρωμόσωμα.
Στις περισσότερες περιπτώσεις (80-85%), ανιχνεύεται ένα φυλοσύνδετο πρότυπο κληρονομικότητας της νόσου, που σχετίζεται με βλάβη στο γονίδιο Col4A5 ως αποτέλεσμα διαγραφής, σημειακών μεταλλάξεων ή διαταραχών συρραφής. Επί του παρόντος, έχουν βρεθεί περισσότερες από 200 μεταλλάξεις του γονιδίου Col4A5, υπεύθυνες για τη διαταραχή της σύνθεσης των α5-αλυσίδων του κολλαγόνου τύπου IV. Με αυτόν τον τύπο κληρονομικότητας, η νόσος εκδηλώνεται σε παιδιά και των δύο φύλων, αλλά στα αγόρια είναι πιο σοβαρή.
Οι μεταλλάξεις στις θέσεις των γονιδίων Col4A3 και Col4A4 που είναι υπεύθυνα για τη σύνθεση των αλυσίδων a3 και a4 του κολλαγόνου τύπου IV κληρονομούνται αυτοσωμικά. Σύμφωνα με την έρευνα, ο αυτοσωμικός επικρατής τύπος κληρονομικότητας παρατηρείται στο 16% των περιπτώσεων κληρονομικής νεφρίτιδας και ο αυτοσωμικός υπολειπόμενος τύπος παρατηρείται στο 6% των ασθενών. Είναι γνωστές περίπου 10 παραλλαγές μεταλλάξεων των γονιδίων Col4A3 και Col4A4.
Το αποτέλεσμα των μεταλλάξεων είναι η παραβίαση των διαδικασιών συναρμολόγησης του κολλαγόνου τύπου IV, που οδηγεί σε παραβίαση της δομής του. Το κολλαγόνο τύπου IV είναι ένα από τα κύρια συστατικά της σπειραματικής βασικής μεμβράνης, της κοχλιακής συσκευής και του φακού του οφθαλμού, η παθολογία της οποίας θα ανιχνευθεί στην κλινική της κληρονομικής νεφρίτιδας.
Το κολλαγόνο τύπου IV, το οποίο αποτελεί μέρος της σπειραματικής βασικής μεμβράνης, αποτελείται κυρίως από δύο αλυσίδες a1 (IV) και μία αλυσίδα a2 (IV), και περιέχει επίσης αλυσίδες a3, a4, a5. Συχνότερα, στην φυλοσύνδετη κληρονομικότητα, η μετάλλαξη του γονιδίου Col4A5 συνοδεύεται από την απουσία αλυσίδων a3, a4, a5 και a6 στη δομή του κολλαγόνου τύπου IV, και ο αριθμός των αλυσίδων o1 και a2 στη σπειραματική βασική μεμβράνη αυξάνεται. Ο μηχανισμός αυτού του φαινομένου είναι ασαφής, θεωρείται ότι η αιτία είναι οι μετα-μεταγραφικές αλλαγές στο mRNA.
Η απουσία αλυσίδων a3, a4 και a5 στη δομή του κολλαγόνου τύπου IV των σπειραματικών βασικών μεμβρανών οδηγεί στην λέπτυνση και την ευθραυστότητά τους στα πρώιμα στάδια του συνδρόμου Alport, το οποίο κλινικά εκδηλώνεται συχνότερα με αιματουρία (λιγότερο συχνά με αιματουρία με πρωτεϊνουρία ή μόνο πρωτεϊνουρία), απώλεια ακοής και φακοειδή σύσπαση. Η περαιτέρω εξέλιξη της νόσου οδηγεί σε πάχυνση και μειωμένη διαπερατότητα των βασικών μεμβρανών στα τελευταία στάδια της νόσου, με τον πολλαπλασιασμό του κολλαγόνου τύπου V και VI σε αυτές, που εκδηλώνεται με αύξηση της πρωτεϊνουρίας και μείωση της νεφρικής λειτουργίας.
Η φύση της μετάλλαξης που αποτελεί τη βάση της κληρονομικής νεφρίτιδας καθορίζει σε μεγάλο βαθμό την φαινοτυπική της εκδήλωση. Στην περίπτωση διαγραφής του χρωμοσώματος Χ με ταυτόχρονη μετάλλαξη των γονιδίων Col4A5 και Col4A6 που είναι υπεύθυνα για τη σύνθεση των α5- και α6-αλυσίδων κολλαγόνου τύπου IV, το σύνδρομο Alport συνδυάζεται με λειομυωμάτωση του οισοφάγου και των γεννητικών οργάνων. Σύμφωνα με ερευνητικά δεδομένα, στην περίπτωση μετάλλαξης του γονιδίου Col4A5 που σχετίζεται με διαγραφή, παρατηρείται μεγαλύτερη σοβαρότητα της παθολογικής διαδικασίας, συνδυασμός νεφρικής βλάβης με εξωνεφρικές εκδηλώσεις και πρώιμη ανάπτυξη χρόνιας νεφρικής ανεπάρκειας, σε σύγκριση με σημειακή μετάλλαξη αυτού του γονιδίου.
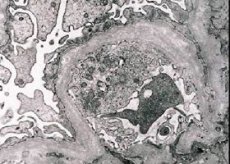
Μορφολογικά, η ηλεκτρονική μικροσκοπία αποκαλύπτει λέπτυνση και στρωματοποίηση των βασικών μεμβρανών των σπειραμάτων (ιδιαίτερα του πυκνού υμένα) και την παρουσία ηλεκτρονιακά πυκνών κοκκίων. Οι σπειραματικές αλλοιώσεις μπορεί να είναι ετερογενείς στον ίδιο ασθενή, από ελάχιστες εστιακές μεσαγγειακές αλλοιώσεις έως σπειραματοσκλήρυνση. Η σπειραματίτιδα στο σύνδρομο Alport είναι πάντα ανοσοαρνητική, γεγονός που τη διακρίνει από τη σπειραματονεφρίτιδα. Χαρακτηριστικά χαρακτηριστικά περιλαμβάνουν την ανάπτυξη σωληναριακής ατροφίας, τη λεμφοϊστιοκυτταρική διήθηση και την παρουσία «αφρωδών κυττάρων» με λιπιδικά εγκλείσματα - λιποφάγα. Καθώς η νόσος εξελίσσεται, αποκαλύπτεται πάχυνση και έντονη καταστροφή των βασικών μεμβρανών των σπειραμάτων.
Αποκαλύπτονται ορισμένες αλλαγές στο ανοσοποιητικό σύστημα. Οι ασθενείς με κληρονομική νεφρίτιδα έχουν μειωμένο επίπεδο IgA και τάση αύξησης της συγκέντρωσης IgM στο αίμα, ενώ το επίπεδο IgG μπορεί να αυξηθεί στα αρχικά στάδια της νόσου και να μειωθεί στα μεταγενέστερα στάδια. Ίσως η αύξηση της συγκέντρωσης IgM και G να είναι ένα είδος αντισταθμιστικής αντίδρασης σε απόκριση στην ανεπάρκεια IgA.
Η λειτουργική δραστηριότητα του συστήματος Τ-λεμφοκυττάρων μειώνεται. παρατηρείται μια επιλεκτική μείωση των Β-λεμφοκυττάρων που είναι υπεύθυνα για τη σύνθεση της IgA, διαταράσσεται η φαγοκυτταρική σύνδεση της ανοσίας, κυρίως λόγω διαταραχής της χημειοταξίας και των ενδοκυτταρικών διεργασιών πέψης στα ουδετερόφιλα.
Κατά την εξέταση βιοψίας νεφρού σε ασθενείς με σύνδρομο Alport, τα δεδομένα ηλεκτρονικής μικροσκοπίας αποκαλύπτουν υπερδομικές αλλαγές στη σπειραματική βασική μεμβράνη: λέπτυνση, διαταραχή της δομής και διάσπαση των σπειραματικών βασικών μεμβρανών με αλλαγή στο πάχος και τα ανώμαλα περιγράμματα. Στα πρώιμα στάδια της κληρονομικής νεφρίτιδας, το ελάττωμα καθορίζει τη λέπτυνση και την ευθραυστότητα των σπειραματικών βασικών μεμβρανών.
Η λέπτυνση των σπειραματικών μεμβρανών είναι ένα πιο ευνοϊκό σημάδι και είναι πιο συχνό στα κορίτσια. Ένα πιο σταθερό ηλεκτρονικό μικροσκοπικό σημάδι στην κληρονομική νεφρίτιδα είναι η διάσπαση της βασικής μεμβράνης, και η σοβαρότητα της καταστροφής της συσχετίζεται με τη σοβαρότητα της διαδικασίας.
Συμπτώματα του συνδρόμου Alport στα παιδιά
Τα πρώτα συμπτώματα του συνδρόμου Alport με τη μορφή μεμονωμένου ουροποιητικού συνδρόμου ανιχνεύονται συχνότερα σε παιδιά των πρώτων τριών ετών ζωής. Στις περισσότερες περιπτώσεις, η ασθένεια ανιχνεύεται τυχαία. Το ουροποιητικό σύνδρομο ανιχνεύεται κατά τη διάρκεια προληπτικής εξέτασης του παιδιού, πριν από την εισαγωγή σε παιδικό σταθμό ή κατά τη διάρκεια ARVI. Σε περίπτωση παθολογίας στα ούρα κατά τη διάρκεια ARVI. Στην κληρονομική νεφρίτιδα, σε αντίθεση με την επίκτητη σπειραματονεφρίτιδα, δεν υπάρχει λανθάνουσα περίοδος.
Στο αρχικό στάδιο της νόσου, η υγεία του παιδιού επηρεάζεται ελάχιστα, χαρακτηριστικό γνώρισμα είναι η επιμονή και η αντοχή του ουροποιητικού συνδρόμου. Ένα από τα κύρια σημάδια είναι η αιματουρία ποικίλου βαθμού σοβαρότητας, που παρατηρείται στο 100% των περιπτώσεων. Αύξηση του βαθμού αιματουρίας παρατηρείται κατά τη διάρκεια ή μετά από αναπνευστικές λοιμώξεις, σωματική δραστηριότητα ή μετά από προληπτικούς εμβολιασμούς. Η πρωτεϊνουρία στις περισσότερες περιπτώσεις δεν υπερβαίνει το 1 g / ημέρα, στην αρχή της νόσου μπορεί να είναι ασταθής, καθώς η διαδικασία εξελίσσεται, η πρωτεϊνουρία αυξάνεται. Περιοδικά, λευκοκυτταρία με κυριαρχία λεμφοκυττάρων μπορεί να υπάρχει στο ίζημα των ούρων, η οποία σχετίζεται με την ανάπτυξη ενδιάμεσων αλλαγών.
Στη συνέχεια, η μερική νεφρική λειτουργία διαταράσσεται, η γενική κατάσταση του ασθενούς επιδεινώνεται: εμφανίζεται μέθη, μυϊκή αδυναμία, αρτηριακή υπόταση, συχνά ακουστική βλάβη (ειδικά στα αγόρια) και μερικές φορές οπτική βλάβη. Η μέθη εκδηλώνεται με ωχρότητα, κόπωση και πονοκεφάλους. Στο αρχικό στάδιο της νόσου, η απώλεια ακοής στις περισσότερες περιπτώσεις ανιχνεύεται μόνο με ακοογραφία. Η απώλεια ακοής στο σύνδρομο Alport μπορεί να εμφανιστεί σε διαφορετικές περιόδους της παιδικής ηλικίας, αλλά συχνότερα η απώλεια ακοής διαγιγνώσκεται στην ηλικία των 6-10 ετών. Η απώλεια ακοής στα παιδιά ξεκινά με υψηλές συχνότητες, φτάνοντας σε σημαντικό βαθμό στην αγωγιμότητα του αέρα και των οστών, περνώντας από την αγωγιμότητα του ήχου στην ηχοαντίληψη της ακοής. Η απώλεια ακοής μπορεί να είναι ένα από τα πρώτα συμπτώματα της νόσου και μπορεί να προηγείται του ουροποιητικού συνδρόμου.
Στο 20% των περιπτώσεων, οι ασθενείς με σύνδρομο Alport παρουσιάζουν αλλαγές στα οπτικά όργανα. Οι πιο συχνά ανιχνεύσιμες ανωμαλίες είναι αυτές του φακού: σφαιροφωκία, πρόσθιος, οπίσθιος ή μικτός φακός και διάφοροι καταρράκτες. Σε οικογένειες με σύνδρομο Alport, υπάρχει σημαντική συχνότητα μυωπίας. Αρκετοί ερευνητές παρατηρούν συνεχώς αμφοτερόπλευρες περιωχρικές αλλαγές σε αυτές τις οικογένειες με τη μορφή φωτεινών υπόλευκων ή κιτρινωπών κοκκιωμάτων στο ωχρό σωμάτιο. Θεωρούν αυτό το σημάδι ως ένα σταθερό σύμπτωμα που έχει υψηλή διαγνωστική αξία στο σύνδρομο Alport. Οι KS Chugh et al. (1993) σε μια οφθαλμολογική μελέτη διαπίστωσαν σε ασθενείς με σύνδρομο Alport μείωση της οπτικής οξύτητας στο 66,7% των περιπτώσεων, πρόσθιο φακό στο 37,8%, κηλίδες στον αμφιβληστροειδή στο 22,2%, καταρράκτη στο 20% και κερατόκωνο στο 6,7%.
Σε ορισμένα παιδιά με κληρονομική νεφρίτιδα, ειδικά όταν αναπτύσσεται νεφρική ανεπάρκεια, παρατηρείται σημαντική υστέρηση στη σωματική ανάπτυξη. Καθώς η νεφρική ανεπάρκεια εξελίσσεται, αναπτύσσεται αρτηριακή υπέρταση. Στα παιδιά, ανιχνεύεται συχνότερα στην εφηβεία και σε μεγαλύτερες ηλικιακές ομάδες.
Οι ασθενείς με κληρονομική νεφρίτιδα χαρακτηρίζονται από την παρουσία διαφόρων (περισσότερων από 5-7) στιγμάτων δυσμορφογένεσης του συνδετικού ιστού. Μεταξύ των στιγμάτων του συνδετικού ιστού στους ασθενείς, τα πιο συνηθισμένα είναι ο υπερτελορισμός των ματιών, η υψηλή υπερώα, οι ανωμαλίες δαγκώματος, το ανώμαλο σχήμα των αυτιών, η καμπυλότητα του μικρού δακτύλου στα χέρια και το "κενό σανδαλιού" στα πόδια. Η κληρονομική νεφρίτιδα χαρακτηρίζεται από την ομοιομορφία των στιγμάτων δυσμορφογένεσης μέσα σε μια οικογένεια, καθώς και από την υψηλή συχνότητα κατανομής τους μεταξύ συγγενών των πιθανών ατόμων από την οποία μεταδίδεται η ασθένεια.
Στα πρώιμα στάδια της νόσου, ανιχνεύεται μια μεμονωμένη μείωση των μερικών νεφρικών λειτουργιών: μεταφορά αμινοξέων, ηλεκτρολυτών, λειτουργία συγκέντρωσης, οξεογένεση, οι μεταγενέστερες αλλαγές επηρεάζουν τη λειτουργική κατάσταση τόσο του εγγύς όσο και του άπω τμήματος του νεφρώνα και χαρακτηρίζονται από συνδυασμένες μερικές διαταραχές. Η μείωση της σπειραματικής διήθησης εμφανίζεται αργότερα, πιο συχνά στην εφηβεία. Καθώς η κληρονομική νεφρίτιδα εξελίσσεται, αναπτύσσεται αναιμία.
Έτσι, η κληρονομική νεφρίτιδα χαρακτηρίζεται από μια σταδιακή πορεία της νόσου: πρώτον, ένα λανθάνον στάδιο ή κρυμμένα κλινικά συμπτώματα, που εκδηλώνονται με ελάχιστες αλλαγές στο ουροποιητικό σύνδρομο, στη συνέχεια εμφανίζεται μια σταδιακή απορύθμιση της διαδικασίας με μείωση της νεφρικής λειτουργίας με εμφανή κλινικά συμπτώματα (δηλητηρίαση, εξασθένιση, αναπτυξιακή καθυστέρηση, αναιμία). Τα κλινικά συμπτώματα εμφανίζονται συνήθως ανεξάρτητα από την στρωματοποίηση της φλεγμονώδους αντίδρασης.
Η κληρονομική νεφρίτιδα μπορεί να εκδηλωθεί σε διαφορετικές ηλικιακές περιόδους, η οποία εξαρτάται από τη δράση του γονιδίου, το οποίο βρίσκεται σε κατασταλμένη κατάσταση μέχρι ένα ορισμένο χρονικό διάστημα.
Ταξινόμηση
Υπάρχουν τρεις τύποι κληρονομικής νεφρίτιδας
- Επιλογή Ι - κλινικά εκδηλώνεται ως νεφρίτιδα με αιματουρία, απώλεια ακοής και οφθαλμική βλάβη. Η πορεία της νεφρίτιδας είναι προοδευτική με την ανάπτυξη χρόνιας νεφρικής ανεπάρκειας. Ο τύπος κληρονομικότητας είναι κυρίαρχος, συνδεδεμένος με το χρωμόσωμα Χ. Μορφολογικά, αποκαλύπτεται παραβίαση της δομής της βασικής μεμβράνης, η λέπτυνση και η σχισμή της.
- Επιλογή II - κλινικά εκδηλώνεται ως νεφρίτιδα με αιματουρία χωρίς απώλεια ακοής. Η πορεία της νεφρίτιδας είναι προοδευτική με την ανάπτυξη χρόνιας νεφρικής ανεπάρκειας. Ο τύπος κληρονομικότητας είναι κυρίαρχος, συνδεδεμένος με το χρωμόσωμα Χ. Μορφολογικά, ανιχνεύεται λέπτυνση της βασικής μεμβράνης των τριχοειδών αγγείων (ιδιαίτερα της ελασματοειδούς).
- Επιλογή III - καλοήθης οικογενής αιματουρία. Η πορεία είναι ευνοϊκή, η χρόνια νεφρική ανεπάρκεια δεν αναπτύσσεται. Ο τύπος κληρονομικότητας είναι αυτοσωμικός κυρίαρχος ή αυτοσωμικός υπολειπόμενος. Με τον αυτοσωμικό υπολειπόμενο τύπο κληρονομικότητας, παρατηρείται μια πιο σοβαρή πορεία της νόσου στις γυναίκες.
Διάγνωση του συνδρόμου Alport
Προτείνονται τα ακόλουθα κριτήρια:
- η παρουσία τουλάχιστον δύο ασθενών με νεφροπάθεια σε κάθε οικογένεια·
- αιματουρία ως το κύριο σύμπτωμα νεφροπάθειας στον υπό εξέταση ασθενή.
- η παρουσία απώλειας ακοής σε τουλάχιστον ένα μέλος της οικογένειας.
- ανάπτυξη χρόνιας νεφρικής ανεπάρκειας σε έναν ή περισσότερους συγγενείς.
Στη διάγνωση διαφόρων κληρονομικών και συγγενών ασθενειών, δίνεται μεγάλη θέση σε μια ολοκληρωμένη προσέγγιση στην εξέταση και, πάνω απ 'όλα, δίνοντας προσοχή στα δεδομένα που λαμβάνονται κατά τη σύνταξη του γενεαλογικού δέντρου του παιδιού. Η διάγνωση του συνδρόμου Alport θεωρείται έγκυρη σε περιπτώσεις όπου ανιχνεύονται 3 από τα 4 τυπικά σημεία στον ασθενή: η παρουσία αιματουρίας και χρόνιας νεφρικής ανεπάρκειας στην οικογένεια, η παρουσία νευροαισθητηριακής απώλειας ακοής, η παθολογία της όρασης στον ασθενή, η ανίχνευση σημείων διάσπασης της σπειραματικής βασικής μεμβράνης με αλλαγή στο πάχος της και ανομοιόμορφα περιγράμματα κατά τη διάρκεια των χαρακτηριστικών της βιοψίας με ηλεκτρονικό μικροσκόπιο.
Η εξέταση του ασθενούς θα πρέπει να περιλαμβάνει κλινικές και γενετικές ερευνητικές μεθόδους, στοχευμένη μελέτη του ιστορικού της νόσου, γενική εξέταση του ασθενούς λαμβάνοντας υπόψη διαγνωστικά σημαντικά κριτήρια. Στο στάδιο της αντιρρόπησης, η παθολογία μπορεί να ανιχνευθεί μόνο εστιάζοντας σε σύνδρομα όπως η παρουσία κληρονομικού φορτίου, η υπόταση, τα πολλαπλά στίγματα δυσεμβρυογένεσης, οι αλλαγές στο ουροποιητικό σύνδρομο. Στο στάδιο της απορύθμισης, μπορεί να εμφανιστούν εξωνεφρικά συμπτώματα, όπως σοβαρή δηλητηρίαση, εξασθένιση, καθυστερημένη σωματική ανάπτυξη, αναιμία, που εκδηλώνονται και εντείνονται με σταδιακή μείωση της νεφρικής λειτουργίας. Στους περισσότερους ασθενείς, με μείωση της νεφρικής λειτουργίας, παρατηρούνται τα ακόλουθα: μειωμένη οξεογένεση και αμινογένεση, το 50% των ασθενών παρατηρούν σημαντική μείωση στην εκκριτική λειτουργία των νεφρών, περιορισμένο εύρος διακυμάνσεων στην οπτική πυκνότητα των ούρων, διαταραχή του ρυθμού διήθησης και στη συνέχεια μείωση της σπειραματικής διήθησης. Το στάδιο της χρόνιας νεφρικής ανεπάρκειας διαγιγνώσκεται όταν οι ασθενείς έχουν αυξημένο επίπεδο ουρίας στον ορό του αίματος (περισσότερο από 0,35 g/l) για 3-6 μήνες ή περισσότερο και μείωση της σπειραματικής διήθησης στο 25% του φυσιολογικού.
Η διαφορική διάγνωση της κληρονομικής νεφρίτιδας θα πρέπει να γίνεται κυρίως με την αιματουρική μορφή της επίκτητης σπειραματονεφρίτιδας. Η επίκτητη σπειραματονεφρίτιδα έχει συχνότερα οξεία έναρξη, περίοδο 2-3 εβδομάδων μετά από μια λοίμωξη, εξωνεφρικά σημεία, συμπεριλαμβανομένης της υπέρτασης από τις πρώτες ημέρες (στην κληρονομική νεφρίτιδα, αντίθετα, υπόταση), μειωμένη σπειραματική διήθηση κατά την έναρξη της νόσου, απουσία διαταραχής των μερικών σωληναριακών λειτουργιών, ενώ στην κληρονομική είναι παρούσες. Η επίκτητη σπειραματονεφρίτιδα εμφανίζεται με πιο έντονη αιματουρία και πρωτεϊνουρία, με αυξημένη ΤΚΕ. Τυπικές αλλαγές στη βασική μεμβράνη των σπειραμάτων, χαρακτηριστικές της κληρονομικής νεφρίτιδας, έχουν διαγνωστική αξία.
Η διαφορική διάγνωση από τη δυσμεταβολική νεφροπάθεια πραγματοποιείται με χρόνια νεφρική ανεπάρκεια, στην οικογένεια κλινικά αποκαλυμμένες ετερογενείς νεφρικές παθήσεις και μπορεί να υπάρχει ένα φάσμα νεφροπάθειας από πυελονεφρίτιδα έως ουρολιθίαση. Τα παιδιά συχνά έχουν παράπονα για πόνο στην κοιλιά και περιοδικά κατά την ούρηση, στα ιζήματα ούρων - οξαλικά.
Εάν υπάρχει υποψία κληρονομικής νεφρίτιδας, ο ασθενής θα πρέπει να παραπεμφθεί σε εξειδικευμένο νεφρολογικό τμήμα για να διευκρινιστεί η διάγνωση.
Τι χρειάζεται να εξετάσετε;
Πώς να εξετάσετε;
Ποιες δοκιμές χρειάζονται;
Ποιος θα επικοινωνήσει;
Θεραπεία του συνδρόμου Alport
Το σχήμα περιλαμβάνει περιορισμούς στην έντονη σωματική άσκηση και την έκθεση στον καθαρό αέρα. Η δίαιτα είναι πλήρης, με επαρκή επίπεδα πλήρων πρωτεϊνών, λιπών και υδατανθράκων, λαμβάνοντας υπόψη τη νεφρική λειτουργία. Μεγάλη σημασία έχει η ανίχνευση και η θεραπεία χρόνιων εστιών λοίμωξης. Χρησιμοποιούνται τα ακόλουθα φάρμακα: ATP, κοκαρβοξυλάση, πυριδοξίνη (έως 50 mg/ημέρα), χλωριούχος καρνιτίνη. Τα μαθήματα χορηγούνται 2-3 φορές το χρόνο. Για την αιματουρία, συνταγογραφείται φυτικό φάρμακο - τσουκνίδα, χυμός αρώνιας, αχίλλεια.
Υπάρχουν αναφορές στην ξένη και εγχώρια βιβλιογραφία σχετικά με τη θεραπεία με πρεδνιζολόνη και τη χρήση κυτταροστατικών. Ωστόσο, είναι δύσκολο να κριθεί το αποτέλεσμα.
Στη χρόνια νεφρική ανεπάρκεια, χρησιμοποιείται αιμοκάθαρση και μεταμόσχευση νεφρού.
Δεν υπάρχουν μέθοδοι ειδικής (αποτελεσματικής παθογενετικής) θεραπείας για την κληρονομική νεφρίτιδα. Όλα τα θεραπευτικά μέτρα στοχεύουν στην πρόληψη και την επιβράδυνση της μείωσης της νεφρικής λειτουργίας.
Η διατροφή πρέπει να είναι ισορροπημένη και υψηλής θερμιδικής αξίας, λαμβάνοντας υπόψη τη λειτουργική κατάσταση των νεφρών. Ελλείψει λειτουργικών διαταραχών, η διατροφή του παιδιού πρέπει να περιέχει επαρκείς πρωτεΐνες, λίπη και υδατάνθρακες. Παρουσία σημείων νεφρικής δυσλειτουργίας, η ποσότητα πρωτεΐνης, υδατανθράκων, ασβεστίου και φωσφόρου πρέπει να είναι περιορισμένη, γεγονός που καθυστερεί την ανάπτυξη χρόνιας νεφρικής ανεπάρκειας.
Η σωματική δραστηριότητα πρέπει να περιορίζεται· τα παιδιά συμβουλεύονται να αποφεύγουν τον αθλητισμό.
Θα πρέπει να αποφεύγεται η επαφή με μολυσματικούς ασθενείς, ο κίνδυνος εμφάνισης οξέων αναπνευστικών νοσημάτων θα πρέπει να μειώνεται. Η απολύμανση των εστιών χρόνιας λοίμωξης είναι απαραίτητη. Δεν πραγματοποιούνται προληπτικοί εμβολιασμοί για παιδιά με κληρονομική νεφρίτιδα, ο εμβολιασμός είναι δυνατός μόνο για επιδημιολογικές ενδείξεις.
Η ορμονική και ανοσοκατασταλτική θεραπεία στην κληρονομική νεφρίτιδα είναι αναποτελεσματική. Υπάρχουν ενδείξεις κάποιας θετικής επίδρασης (μείωση της πρωτεϊνουρίας και επιβράδυνση της εξέλιξης της νόσου) με τη μακροχρόνια πολυετή χρήση κυκλοσπορίνης Α και αναστολέων ΜΕΑ.
Στη θεραπεία των ασθενών, χρησιμοποιούνται φάρμακα που βελτιώνουν τον μεταβολισμό:
- πυριδοξίνη - 2-3 mg/kg/ημέρα σε 3 δόσεις για 4 εβδομάδες.
- κοκαρβοξυλάση - 50 mg ενδομυϊκά κάθε δεύτερη ημέρα, συνολικά 10-15 ενέσεις.
- ATP - 1 ml ενδομυϊκά κάθε δεύτερη ημέρα, 10-15 ενέσεις.
- βιταμίνη Α - 1000 IU/έτος/ημέρα σε 1 δόση για 2 εβδομάδες.
- Βιταμίνη Ε - 1 mg/kg/ημέρα σε 1 δόση για 2 εβδομάδες.
Αυτός ο τύπος θεραπείας βοηθά στη βελτίωση της γενικής κατάστασης των ασθενών, στη μείωση των σωληναριακών δυσλειτουργιών και πραγματοποιείται σε μαθήματα 3 φορές το χρόνο.
Η λεβαμισόλη μπορεί να χρησιμοποιηθεί ως ανοσοτροποποιητής - 2 mg/kg/ημέρα 2-3 φορές την εβδομάδα με διαλείμματα 3-4 ημερών μεταξύ των δόσεων.
Σύμφωνα με ερευνητικά δεδομένα, η υπερβαρική οξυγόνωση έχει θετική επίδραση στη σοβαρότητα της αιματουρίας και της νεφρικής δυσλειτουργίας.
Η πιο αποτελεσματική μέθοδος αντιμετώπισης της κληρονομικής νεφρίτιδας είναι η έγκαιρη μεταμόσχευση νεφρού. Σε αυτή την περίπτωση, δεν υπάρχει υποτροπή της νόσου στο μόσχευμα. Σε ένα μικρό ποσοστό περιπτώσεων (περίπου 5%), μπορεί να αναπτυχθεί νεφρίτιδα στο μεταμοσχευμένο νεφρό που σχετίζεται με αντιγόνα στη βασική μεμβράνη του σπειράματος.
Μια πολλά υποσχόμενη κατεύθυνση είναι η προγεννητική διάγνωση και η γενετική μηχανική θεραπεία. Πειράματα σε ζώα δείχνουν υψηλή αποτελεσματικότητα στη μεταφορά φυσιολογικών γονιδίων που είναι υπεύθυνα για τη σύνθεση αλυσίδων κολλαγόνου τύπου IV στον νεφρικό ιστό, μετά την οποία παρατηρείται η σύνθεση φυσιολογικών δομών κολλαγόνου.
Πρόβλεψη
Η πρόγνωση για την κληρονομική νεφρίτιδα είναι πάντα σοβαρή.
Τα προγνωστικά δυσμενή κριτήρια για την πορεία της κληρονομικής νεφρίτιδας είναι:
- αρσενικό φύλο;
- πρώιμη ανάπτυξη χρόνιας νεφρικής ανεπάρκειας σε μέλη της οικογένειας.
- πρωτεϊνουρία (περισσότερο από 1 g/ημέρα)
- πάχυνση των σπειραματικών βασικών μεμβρανών σύμφωνα με τη μικροσκοπία.
- ακουστική νευρίτιδα;
- διαγραφή στο γονίδιο Col4A5.
Η πρόγνωση για καλοήθη οικογενή αιματουρία είναι πιο ευνοϊκή.
Использованная литература