Ιατρικός εμπειρογνώμονας του άρθρου

Νέες δημοσιεύσεις
Κερατόδερμα: αιτίες, συμπτώματα, διάγνωση, θεραπεία
Τελευταία επισκόπηση: 07.07.2025

Όλα τα περιεχόμενα του iLive ελέγχονται ιατρικά ή ελέγχονται για να διασφαλιστεί η όσο το δυνατόν ακριβέστερη ακρίβεια.
Έχουμε αυστηρές κατευθυντήριες γραμμές προμήθειας και συνδέουμε μόνο με αξιόπιστους δικτυακούς τόπους πολυμέσων, ακαδημαϊκά ερευνητικά ιδρύματα και, όπου είναι δυνατόν, ιατρικά επισκοπικά μελέτες. Σημειώστε ότι οι αριθμοί στις παρενθέσεις ([1], [2], κλπ.) Είναι σύνδεσμοι με τις οποίες μπορείτε να κάνετε κλικ σε αυτές τις μελέτες.
Εάν πιστεύετε ότι κάποιο από το περιεχόμενό μας είναι ανακριβές, παρωχημένο ή αμφισβητήσιμο, παρακαλώ επιλέξτε το και πατήστε Ctrl + Enter.
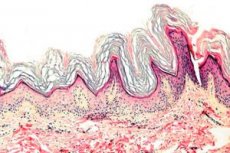
Η κερατοδερμία είναι μια ομάδα δερματοπαθειών που χαρακτηρίζονται από διαταραχή της διαδικασίας κερατινοποίησης - υπερβολικό σχηματισμό κερατοειδούς κυρίως στις παλάμες και τα πέλματα.
Τα αίτια και η παθογένεση της νόσου δεν έχουν διευκρινιστεί πλήρως. Η έρευνα έχει αποδείξει ότι τα κερατοδερμικά προκαλούνται από μεταλλάξεις στα γονίδια που κωδικοποιούν την κερατίνη 6, 9, 16. Η ανεπάρκεια βιταμίνης Α, οι ορμονικές δυσλειτουργίες, κυρίως των γεννητικών αδένων, οι βακτηριακές και ιογενείς λοιμώξεις έχουν μεγάλη σημασία στην παθογένεση. Αποτελούν ένα από τα συμπτώματα κληρονομικών ασθενειών και όγκων εσωτερικών οργάνων (παραψωριασικά κερατοδερμικά).
Συμπτώματα. Γίνεται διάκριση μεταξύ διάχυτης (κερατόδερμα Unna-Tost, κερατόδερμα Meleda, κερατόδερμα Papillon-Lefevre, ακρωτηριαστική κερατόδερμα και σύνδρομα που περιλαμβάνουν τη διάχυτη κερατόδερμα ως ένα από τα κύρια συμπτώματα) και εστιακής (διάχυτη κηλιδωτή κερατόδερμα Fischer-Buschke, ακροκερατοελαστοείδωση Kosti, περιορισμένη κερατόδερμα Bruhauer-Franzesthesti, γραμμική κερατόδερμα Fuchs, κ.λπ.) κερατόδερμα.
Η κερατοδερμία Winy-Tost (συνώνυμα: συγγενής ιχθύωση των παλαμών και των πελμάτων, σύνδρομο Winy-Tost) μεταδίδεται με αυτοσωμικό κυρίαρχο τρόπο. Υπάρχει μια διάχυτη υπερβολική κερατινοποίηση του δέρματος των παλαμών και των πελμάτων (μερικές φορές μόνο των πελμάτων), η οποία αναπτύσσεται κατά τα πρώτα δύο χρόνια της ζωής. Η παθολογική διαδικασία του δέρματος ξεκινά με μια ελαφρά πάχυνση του δέρματος των παλαμών και των πελμάτων με τη μορφή μιας λωρίδας ερυθήματος με ωχρό χρώμα στα όρια με το υγιές δέρμα. Με την πάροδο του χρόνου, εμφανίζονται λείες, κιτρινωπές κερατώδεις στρώσεις στην επιφάνειά τους. Η βλάβη σπάνια εξαπλώνεται στη ράχη των καρπών ή των δακτύλων. Σε ορισμένους ασθενείς, μπορεί να σχηματιστούν επιφανειακές ή βαθιές ρωγμές και παρατηρείται τοπική υπεριδρωσία. Στον ασθενή που παρατηρείται από τον συγγραφέα, ο θείος από την πλευρά της μητέρας, ο αδελφός και ο γιος έπασχαν από κερατοδερμία Winy-Tost.
Περιγράφονται περιπτώσεις βλάβης στα νύχια (πάχυνση), τα δόντια και τα μαλλιά στην κερατοδερμία Winy-Tost σε συνδυασμό με διάφορες σκελετικές ανωμαλίες και παθολογίες εσωτερικών οργάνων, νευρικού και ενδοκρινικού συστήματος.
Ιστοπαθολογία. Η ιστολογική εξέταση αποκαλύπτει έντονη υπερκεράτωση, κοκκίωση, ακάνθωση και μικρές φλεγμονώδεις διηθήσεις στο άνω χόριο. Διαφορική διάγνωση. Η νόσος πρέπει να διαφοροδιαγνωστεί από άλλους τύπους κερατοδερμίας.
Η κερατοδερμία Meleda (συνώνυμα: νόσος Meleda, συγγενές προοδευτικό ακροκεράτωμα, παλαμοπελματιαία διακλαδική κεράτωση Siemens, κληρονομική παλαμοπελματιαία προοδευτική κεράτωση Kogoy) κληρονομείται με αυτοσωμικό υπολειπόμενο τρόπο. Αυτή η μορφή κερατοδερμίας χαρακτηρίζεται από παχιά, κιτρινοκαφέ κερατώδη στρώματα με βαθιές ρωγμές. Ένα βιολετί-μωβ περίγραμμα πλάτους αρκετών χιλιοστών είναι ορατό κατά μήκος των άκρων της βλάβης. Η εξεργασία συνήθως εξαπλώνεται στο πίσω μέρος των χεριών και των ποδιών, στους αντιβράχιους και στις κνήμες. Οι περισσότεροι ασθενείς εμφανίζουν τοπική υπεριδρωσία. Από αυτή την άποψη, η επιφάνεια των παλαμών και των πελμάτων γίνεται ελαφρώς υγρή και καλύπτεται με μαύρες κουκκίδες (πόροι ιδρωτοποιών αδένων).
Η ασθένεια μπορεί να αναπτυχθεί μέχρι την ηλικία των 15-20 ετών. Τα νύχια παχύνονται και παραμορφώνονται.
Ιστοπαθολογία. Η ιστολογική εξέταση αποκαλύπτει υπερκεράτωση, μερικές φορές ακάνθωση, και χρόνια φλεγμονώδη διήθηση στο θηλώδες χόριο.
Διαφορική διάγνωση. Η κερατοδερμία Melela πρέπει να διακρίνεται από την κερατοδερμία Unna-Tost.
Το κερατόδερμα Papillon-Lefevre (συνώνυμο: παλαμοπλαντική υπερκεράτωση με περιοδοντίτιδα) κληρονομείται με αυτοσωμικό υπολειπόμενο τρόπο.
Η νόσος εκδηλώνεται στο 2ο-3ο έτος της ζωής. Η κλινική εικόνα της νόσου είναι παρόμοια με τη νόσο του Μελέλα. Επιπλέον, οι αλλαγές στα δόντια είναι χαρακτηριστικές (ανωμαλίες στην ανατολή των γαλακτοφόρων και μόνιμων δοντιών με την ανάπτυξη τερηδόνας, ουλίτιδα, ταχέως εξελισσόμενη περιοδοντίτιδα με πρόωρη απώλεια δοντιών).
Ιστοπαθολογία. Η ιστολογική εξέταση αποκαλύπτει πάχυνση όλων των στιβάδων της επιδερμίδας, ειδικά της κερατώδους στιβάδας, και ασήμαντες κυτταρικές συστάδες λεμφοκυττάρων και ιστιοκυττάρων στο χόριο.
Διαφορική διάγνωση. Η νόσος θα πρέπει να διακρίνεται από άλλα κερατοδερμικά. Ένα σημαντικό διακριτικό χαρακτηριστικό είναι η χαρακτηριστική οδοντική παθολογία, η οποία δεν απαντάται σε άλλες μορφές κληρονομικών διάχυτων κερατοδερμικών.
Το κερατόδερμα mutilans (συνώνυμα: σύνδρομο Fonwinkel, κληρονομικό ακρωτηριαστικό κερατώμα) είναι ένας τύπος διάχυτης κερατοδερμίας που κληρονομείται με αυτοσωμικό επικρατή τρόπο. Αναπτύσσεται στο 2ο έτος της ζωής και χαρακτηρίζεται από διάχυτες κερατώδεις εναποθέσεις στο δέρμα των παλαμών και των πελμάτων με υπεριδρωσία. Με την πάροδο του χρόνου, σχηματίζονται αυλακώσεις που μοιάζουν με χορδές στα δάχτυλα, οι οποίες οδηγούν σε συσπάσεις και αυθόρμητο ακρωτηριασμό των δακτύλων. Η θυλακοειδής κεράτωση εκφράζεται στο πίσω μέρος των χεριών, καθώς και στην περιοχή των αρθρώσεων του αγκώνα και του γονάτου. Οι πλάκες των νυχιών αλλοιώνονται (συχνά σαν γυαλιά ρολογιού). Έχουν περιγραφεί περιπτώσεις υπογοναδισμού, αλωπεκίας ρουμπινιού, απώλειας ακοής, παχυονυχίας.
Ιστοπαθολογία. Η ιστολογική εξέταση αποκαλύπτει σοβαρή υπερκεράτωση, κοκκίωση, ακάνθωση και μικρές φλεγμονώδεις διηθήσεις στο χόριο, που αποτελούνται από λεμφοκύτταρα και ιστιοκύτταρα.
Διαφορική διάγνωση. Κατά τη διαφοροποίηση της ακρωτηριαστικής κερατοδερμίας από άλλες μορφές διάχυτης κερατοδερμίας, θα πρέπει πρώτα απ 'όλα να λαμβάνεται υπόψη η επίδραση του ακρωτηριασμού, η οποία δεν είναι τυπική για άλλες μορφές. Κατά τη διενέργεια διαφορικής διάγνωσης όλων των μορφών διάχυτης κερατοδερμίας, είναι απαραίτητο να θυμόμαστε ότι μπορεί να είναι ένα από τα κύρια συμπτώματα μιας σειράς κληρονομικών συνδρόμων.
Θεραπεία. Η νεοτιγαζόνη ενδείκνυται στη γενική θεραπεία της κερατοδερμίας. Η δόση του φαρμάκου εξαρτάται από τη σοβαρότητα της διαδικασίας και είναι 0,3-1 mg/kg του βάρους του ασθενούς. Σε περίπτωση απουσίας νεοτιγαζόνης, συνιστάται η χορήγηση βιταμίνης Α σε δόση 100 έως 300.000 mg την ημέρα για μεγάλο χρονικό διάστημα. Η εξωτερική θεραπεία συνίσταται στη χρήση αλοιφών με αρωματικά ρετινοειδή, κερατολυτικούς και στεροειδή παράγοντες.
 [
[ Τι σε προβληματιζει?
Τι χρειάζεται να εξετάσετε;
Πώς να εξετάσετε;