Ιατρικός εμπειρογνώμονας του άρθρου

Νέες δημοσιεύσεις
Σύνδρομο Angelman σε παιδιά και ενήλικες
Τελευταία επισκόπηση: 04.07.2025

Όλα τα περιεχόμενα του iLive ελέγχονται ιατρικά ή ελέγχονται για να διασφαλιστεί η όσο το δυνατόν ακριβέστερη ακρίβεια.
Έχουμε αυστηρές κατευθυντήριες γραμμές προμήθειας και συνδέουμε μόνο με αξιόπιστους δικτυακούς τόπους πολυμέσων, ακαδημαϊκά ερευνητικά ιδρύματα και, όπου είναι δυνατόν, ιατρικά επισκοπικά μελέτες. Σημειώστε ότι οι αριθμοί στις παρενθέσεις ([1], [2], κλπ.) Είναι σύνδεσμοι με τις οποίες μπορείτε να κάνετε κλικ σε αυτές τις μελέτες.
Εάν πιστεύετε ότι κάποιο από το περιεχόμενό μας είναι ανακριβές, παρωχημένο ή αμφισβητήσιμο, παρακαλώ επιλέξτε το και πατήστε Ctrl + Enter.

Υπάρχουν ορισμένες ασθένειες για τις οποίες εκφράσεις όπως «φρόντισε τον εαυτό σου και δεν θα αρρωστήσεις» ακούγονται, τουλάχιστον, γελοίες. Πρόκειται για παθολογίες στις οποίες ορισμένες ψυχικές και σωματικές ανωμαλίες είναι εγγενείς στο σώμα του παιδιού ακόμη και πριν από τη γέννηση, αλλά οι γονείς δεν φταίνε γι' αυτό. Τέτοιες ασθένειες προκαλούνται από μεταλλάξεις ή ανωμαλίες σε σύνολα χρωμοσωμάτων και ονομάζονται χρωμοσωμικές ή γενετικές. Σύνδρομο Angelman, σύνδρομο Down, σύνδρομο Patau, σύνδρομο Edwards, σύνδρομο Turner, σύνδρομο Prader-Willi - αυτό είναι μόνο ένα μέρος των γενετικών ασθενειών από μια αρκετά αξιοπρεπή λίστα.
Σύνδρομο Ευτυχισμένου Ανθρώπου
Αυτή τη φορά θα μιλήσουμε για την παθολογία που πήρε το όνομά της από τον Άγγλο παιδίατρο Χάρι Άντζελμαν, ο οποίος έθεσε για πρώτη φορά το ζήτημα αυτού του προβλήματος το 1965, έχοντας συναντήσει τρία ασυνήθιστα παιδιά στο ιατρείο του την προηγούμενη μέρα, τα οποία ένωναν κοινά, ιδιαίτερα συμπτώματα. Ο γιατρός ονόμασε αυτά τα παιδιά παιδιά-κούκλες και έγραψε ένα άρθρο γι' αυτά, το οποίο αρχικά ονομαζόταν "Παιδιά-μαριονέτες". Το ίδιο το άρθρο και ο τίτλος του γράφτηκαν με την εντύπωση ενός πίνακα που βρισκόταν σε ένα από τα μουσεία της Βερόνα. Ο πίνακας απεικόνιζε ένα γελαστό αγόρι και ονομαζόταν "Το Αγόρι-Μαριονέτα". Η συσχέτιση του παιδιού που απεικονίζεται στον πίνακα με τα τρία παιδιά που συνάντησε κάποτε ο Άντζελμαν στο ιατρείο του ώθησε τον παιδίατρο να συνδυάσει τα παιδιά σε μία ομάδα λόγω της ασθένειας που είχαν.
Δεν υπάρχει τίποτα το περίεργο στο γεγονός ότι τα παιδιά που αναφέρονται στο άρθρο δεν έγιναν αντιληπτά από άλλους γιατρούς. Άλλωστε, με την πρώτη ματιά φαινόταν ότι είχαν εντελώς διαφορετικές ασθένειες, τόσο διαφορετική ήταν η γενική κλινική εικόνα της νόσου σε 3 διαφορετικές περιπτώσεις. Ίσως η «νέα» χρωμοσωμική παθολογία να ενδιέφερε άλλους επιστήμονες, αλλά εκείνη την εποχή η γενετική δεν είχε ακόμη αναπτυχθεί αρκετά για να επιβεβαιώσει την υπόθεση του Άγγλου γιατρού. Επομένως, μετά από ένα ορισμένο ενδιαφέρον για αυτήν, το άρθρο πετάχτηκε στο πίσω ράφι για μεγάλο χρονικό διάστημα.
Η επόμενη αναφορά στο σύνδρομο Angelman, όπως ονομάστηκε τώρα το άρθρο του Άγγλου παιδίατρου G. Angelman, χρονολογείται στις αρχές της δεκαετίας του '80 του 20ού αιώνα. Και μόνο το 1987 ήταν δυνατό να βρεθεί ο λόγος για τον οποίο ένα μικρό μέρος των παιδιών γεννιέται με τέτοιες αποκλίσεις που από έξω φαίνονται να χαμογελούν συνεχώς και να είναι χαρούμενα. Στην πραγματικότητα, αυτό δεν είναι καθόλου αλήθεια, και το χαμόγελο είναι απλώς μια γκριμάτσα, πίσω από την οποία κρύβεται μια δυστυχισμένη ανθρώπινη ψυχή και ο πόνος των γονέων.
Επιδημιολογία
Σύμφωνα με στατιστικά στοιχεία, μια χρωμοσωμική μετάλλαξη σε ένα παιδί μπορεί να αναπτυχθεί τόσο στο πλαίσιο παρόμοιων μεταλλάξεων στους γονείς όσο και ελλείψει τέτοιων. Δεν υπάρχει σαφής κληρονομική φύση του συνδρόμου Angelman (AS), αλλά η πιθανότητα εμφάνισης παθολογίας σε γονείς με χρωμοσωμικές μεταλλάξεις είναι αρκετά υψηλή.
Είναι επίσης ενδιαφέρον ότι εάν μια οικογένεια έχει ήδη ένα παιδί με Σύνδρομο Άσπεργκερ, υπάρχει ένα τοις εκατό πιθανότητα να αποκτήσει ένα δεύτερο παιδί με την ίδια διαταραχή, ακόμη και αν οι γονείς είναι υγιείς.
Δεν υπάρχουν ακόμη ακριβή στατιστικά στοιχεία για τον αριθμό των ασθενών με σύνδρομο Angelman. Ίσως ο λόγος είναι η ποικιλία των συμπτωμάτων, τα οποία μπορεί να εμφανιστούν σε μια συγκεκριμένη σύνθεση ή να μην εμφανιστούν καθόλου για μεγάλο χρονικό διάστημα. Υποτίθεται ότι η συχνότητα εμφάνισης της νόσου είναι: 1 παιδί ανά 20.000 νεογνά. Αλλά αυτός ο αριθμός είναι πολύ κατά προσέγγιση.
Αιτίες Σύνδρομο Angelman
Το σύνδρομο Angelman είναι μια ιατρική ονομασία για μια χρωμοσωμική παθολογία, αλλά δεν είναι η μόνη. Οι άνθρωποι αποκαλούν αυτήν την ασθένεια σύνδρομο των παιδιών-κουκλών, σύνδρομο χαρούμενης μαριονέτας, σύνδρομο Petrushka και σύνδρομο γελαστής κούκλας. Οι άνθρωποι βρίσκουν κάθε είδους ονόματα (μερικές φορές ακόμη και προσβλητικά για τους ίδιους τους ασθενείς και τους γονείς τους), αλλά μια ασθένεια είναι μια ασθένεια, όσο αστεία κι αν φαίνεται και ανεξάρτητα από τις αιτίες.
Και οι λόγοι για την ανάπτυξη του συνδρόμου Angelman, όπως και πολλών άλλων γενετικών παθολογιών, σε όλες τις περιπτώσεις είναι οι διαταραχές στη δομή ενός από τα χρωμοσώματα ή του συνόλου των χρωμοσωμάτων στο σύνολό τους. Αλλά στην περίπτωσή μας, όλο το πρόβλημα έγκειται στο χρωμόσωμα 15, που μεταδίδεται από τη μητέρα. Δηλαδή, το πατρικό χρωμόσωμα σε αυτή την περίπτωση δεν έχει αποκλίσεις, αλλά το θηλυκό υφίσταται ορισμένες μεταλλάξεις.
Ανάλογα με τον τύπο της χρωμοσωμικής ανωμαλίας, το σύνδρομο Angelman ταξινομείται ως χρωμοσωμική μετάλλαξη. Τέτοιες μεταλλάξεις θεωρούνται:
- Μια διαγραφή (απουσία ενός τμήματος ενός χρωμοσώματος που περιέχει ένα συγκεκριμένο σύνολο γονιδίων· εάν ένα από τα γονίδια λείπει, μιλάμε για μικροδιαγραφή), η οποία είναι το αποτέλεσμα δύο θραύσεων και μίας επανένωσης, όταν χάνεται ένα τμήμα του αρχικού χρωμοσώματος.
- Διπλασιασμός (η παρουσία ενός επιπλέον τμήματος σε ένα χρωμόσωμα που είναι αντίγραφο ενός υπάρχοντος), ο οποίος στις περισσότερες περιπτώσεις οδηγεί στον θάνατο ενός ατόμου και λιγότερο συχνά σε υπογονιμότητα.
- Αναστροφή (αναστροφή ενός από τα τμήματα του χρωμοσώματος κατά 180 μοίρες, δηλαδή προς την αντίθετη κατεύθυνση, και στη συνέχεια τα γονίδια σε αυτό βρίσκονται στην αντίθετη σειρά), όταν τα σπασμένα άκρα του χρωμοσώματος συνδέονται με διαφορετική σειρά από την αρχική.
- Εισαγωγή (εάν μέρος του γενετικού υλικού σε ένα χρωμόσωμα είναι εκτός θέσης),
- μετατόπιση (εάν ένα συγκεκριμένο τμήμα ενός χρωμοσώματος συνδέεται με ένα άλλο χρωμόσωμα· μια τέτοια μετάλλαξη μπορεί να είναι αμοιβαία χωρίς απώλεια τμημάτων).
Λαμβάνοντας ένα μεταλλαγμένο χρωμόσωμα από μια ανυποψίαστη μητέρα, το παιδί είναι καταδικασμένο να γεννηθεί με ανωμαλίες. Η πιο συχνή αιτία του συνδρόμου Angelman εξακολουθεί να θεωρείται η διαγραφή του μητρικού 15ου χρωμοσώματος, όταν λείπει ένα μικρό τμήμα. Λιγότερο συχνές μεταλλάξεις στο σύνδρομο της «γελαστής κούκλας» θεωρούνται οι εξής:
- μετατόπιση,
- μονοπατρική δισωμία (εάν το παιδί έλαβε ένα ζεύγος χρωμοσωμάτων από τον πατέρα, το μητρικό χρωμόσωμα απουσιάζει),
- μετάλλαξη γονιδίων στο DNA, τα οποία αποτελούν τόσο το κύριο δομικό (γενετικό) υλικό όσο και οδηγίες για τη σωστή χρήση του (ιδιαίτερα, μετάλλαξη του γονιδίου ube3a στο μητρικό χρωμόσωμα).
Η παρουσία μίας από αυτές τις μεταλλάξεις στους γονείς αποτελεί παράγοντα κινδύνου για την ανάπτυξη του συνδρόμου Angelman στα παιδιά. Αλλά όχι μόνο οι χρωμοσωμικές μεταλλάξεις, αλλά και οι γονιδιωματικές (οι οποίες σχετίζονται με μια ποσοτική αλλαγή στα σύνολα χρωμοσωμάτων και είναι πιο συχνές από τις χρωμοσωμικές) μπορούν να προκαλέσουν την ανάπτυξη της νόσου σε ένα παιδί. Οι συχνές γονιδιωματικές μεταλλάξεις περιλαμβάνουν την τρισωμία χρωμοσωμάτων (εάν το σύνολο χρωμοσωμάτων ενός ατόμου έχει περισσότερα από 46 χρωμοσώματα).
Για να εμφανιστεί μια παθολογία σε ένα παιδί, δεν είναι καθόλου απαραίτητο οι γονείς να έχουν χρωμοσωμικές ανωμαλίες. Κι όμως, υπάρχει ένα ορισμένο ποσοστό ασθενών των οποίων η νόσος είναι κληρονομική.
Παθογένεση
Ας εμβαθύνουμε λίγο περισσότερο στη βιολογία, ή πιο συγκεκριμένα, στη γενετική. Οι γενετικές πληροφορίες κάθε ανθρώπινου οργανισμού περιέχονται σε 23 ζεύγη χρωμοσωμάτων. Το ένα χρωμόσωμα από ένα ζεύγος μεταβιβάζεται στο παιδί από τον πατέρα και το άλλο από τη μητέρα. Όλα τα ζεύγη χρωμοσωμάτων διαφέρουν σε σχήμα και μέγεθος και φέρουν ορισμένες πληροφορίες. Έτσι, το 23ο ζεύγος χρωμοσωμάτων (χρωμοσώματα Χ και Υ) είναι υπεύθυνο για τον σχηματισμό των σεξουαλικών χαρακτηριστικών του μωρού (XX - κορίτσι, XY - αγόρι, ενώ το χρωμόσωμα Υ μπορεί να ληφθεί από το παιδί μόνο από τον πατέρα).
Ιδανικά, ένα παιδί λαμβάνει 46 χρωμοσώματα από τους γονείς του, τα οποία σχηματίζουν τα γενετικά του χαρακτηριστικά, προκαθορίζοντάς το ως άτομο. Ένας μεγαλύτερος αριθμός χρωμοσωμάτων ονομάζεται τρισωμία και θεωρείται απόκλιση από τον κανόνα. Για παράδειγμα, η παρουσία του χρωμοσώματος 47 στο σύνολο χρωμοσωμάτων (καρυότυπος, προσδιορισμός είδους και ατομικών χαρακτηριστικών) προκαλεί την εμφάνιση συνδρόμου Down.
Εάν τα χρωμοσώματα χρωματιστούν με μια ειδική χρωστική ουσία, τότε κάτω από το μικροσκόπιο μπορείτε να δείτε ρίγες διαφορετικών αποχρώσεων κατά μήκος καθεμίας από αυτές. Μέσα σε κάθε λωρίδα υπάρχει ένας τεράστιος αριθμός γονιδίων. Όλες αυτές οι ρίγες αριθμούνται από τους επιστήμονες και έχουν μια σταθερή θέση. Η απουσία μιας από τις ρίγες θεωρείται απόκλιση από τον κανόνα. Στο σύνδρομο Angelman, μπορεί κανείς πολύ συχνά να παρατηρήσει την απουσία τμημάτων του μητρικού χρωμοσώματος στο διάστημα q11-q13, που βρίσκεται στον μακρύ βραχίονα, ο αριθμός των βάσεων DNA στο οποίο είναι μόνο περίπου 4 εκατομμύρια.
Το κύριο συστατικό του χρωμοσώματος θεωρείται ένα απίστευτα μακρύ μόριο DNA που περιέχει χιλιάδες γονίδια και δεκάδες και εκατοντάδες εκατομμύρια αζωτούχες βάσεις. Έτσι, το χρωμόσωμα 15, υπεύθυνο για την ανάπτυξη του συνδρόμου Angelman και αρκετών άλλων, περιέχει 1200 γονίδια και περίπου 100 εκατομμύρια βάσεις. Οποιεσδήποτε διαταραχές στη δομή του μορίου DNA σίγουρα θα επηρεάσουν την εμφάνιση και την ανάπτυξη του μελλοντικού παιδιού.
Οι γενετικές πληροφορίες που περιέχονται στα γονίδια μετατρέπονται σε πρωτεΐνη ή RNA. Αυτή η διαδικασία ονομάζεται γονιδιακή έκφραση. Με αυτόν τον τρόπο, οι γενετικές πληροφορίες που λαμβάνονται από τους γονείς λαμβάνουν τόσο μορφή όσο και περιεχόμενο, το οποίο ενσωματώνεται στον μοναδικό θηλυκό ή αρσενικό κληρονόμο τους.
Υπάρχουν ορισμένες παθολογίες με μη κλασικό τύπο κληρονομικότητας, συμπεριλαμβανομένου του συνδρόμου Angelman, στο οποίο τα γονίδια που λαμβάνονται από τους γονείς ως μέρος ζευγαρωμένων χρωμοσωμάτων φέρουν ένα μοναδικό αποτύπωμα των γονέων και εκδηλώνονται με διαφορετικούς τρόπους.
Έτσι, το σύνδρομο Angelman είναι ένα εντυπωσιακό παράδειγμα γονιδιωματικής αποτύπωσης, στο οποίο η γονιδιακή έκφραση στο σώμα του παιδιού εξαρτάται άμεσα από τον γονέα από τον οποίο ελήφθησαν τα αλληλόμορφα (διαφορετικές μορφές ενός γονιδίου, που ελήφθησαν από τον πατέρα και τη μητέρα, που βρίσκονται σε πανομοιότυπα τμήματα ζευγαρωμένων χρωμοσωμάτων). Δηλαδή, μόνο ανωμαλίες στο μητρικό χρωμόσωμα οδηγούν στην ανάπτυξη του συνδρόμου, ενώ μεταλλάξεις και δομικές διαταραχές του πατρικού χρωμοσώματος προκαλούν εντελώς διαφορετικές παθολογίες.
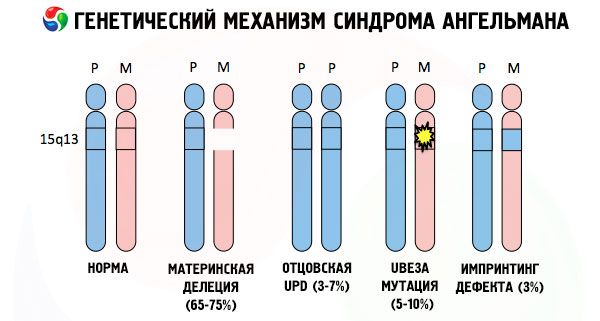
Σε αυτή την παθολογία, υπάρχει έλλειψη ορισμένων γονιδίων στο μητρικό χρωμόσωμα ή απώλεια/μείωση της δραστηριότητας μεμονωμένων γονιδίων (στη συντριπτική πλειονότητα των περιπτώσεων, του γονιδίου ube3a, το οποίο εμπλέκεται στον μεταβολισμό της ουβικιτίνης, μιας πρωτεΐνης που ρυθμίζει την αποικοδόμηση άλλων πρωτεϊνών). Ως αποτέλεσμα, το παιδί διαγιγνώσκεται με νοητικές αναπτυξιακές ανωμαλίες και σωματικές παραμορφώσεις.
Συμπτώματα Σύνδρομο Angelman
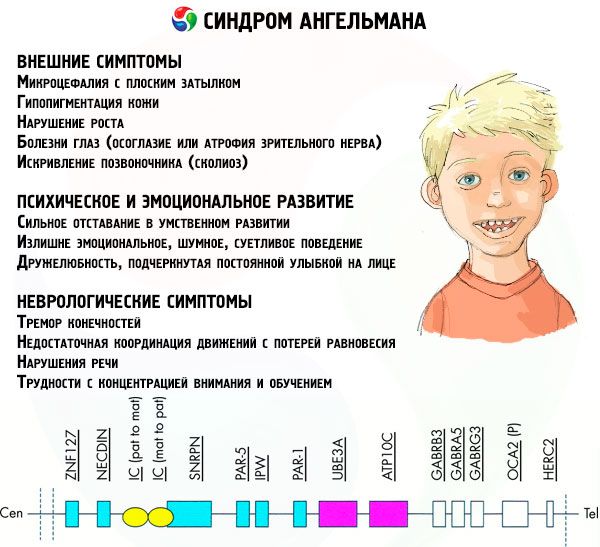
Τα συμπτώματα του συνδρόμου Angelman επηρεάζουν διάφορες πτυχές της ζωής και της ανάπτυξης ενός παιδιού: σωματική, νευρολογική, ψυχική. Με βάση αυτό, μπορούν να εντοπιστούν 3 ομάδες συμπτωμάτων που υποδηλώνουν την ανάπτυξη αυτής της παθολογίας.
- Εξωτερικά ή σωματικά συμπτώματα:
- δυσανάλογα μικρό κεφάλι σε σύγκριση με το σώμα και τα άκρα, τα οποία έχουν φυσιολογικό μέγεθος,
- πολύ πλατύ στόμα,
- σχεδόν πάντα υπάρχει ένα χαμόγελο στο πρόσωπο (με ανοιχτό στόμα),
- αραιά δόντια,
- στενό άνω χείλος,
- συχνά προεξέχουσα πλατιά γλώσσα,
- προεξέχουσα κάτω γνάθος,
- μυτερό πηγούνι,
- πολύ ανοιχτόχρωμο δέρμα, συχνά τρίχωμα (αλβινισμός, που σχετίζεται με το γεγονός ότι το σώμα δεν παράγει τη χρωστική μελανίνη),
- σκούρες κηλίδες σε ανοιχτόχρωμο δέρμα (υπομελάγχρωση λόγω ανεπαρκούς παραγωγής μελανίνης)
- σωματικά ή εξωτερικά συμπτώματα: οφθαλμικές παθήσεις όπως στραβισμός ή ατροφία οπτικού νεύρου,
- καμπυλότητα της σπονδυλικής στήλης (σκολίωση),
- δύσκαμπτα πόδια (όταν περπατάει, ένα άτομο δεν λυγίζει τα πόδια του στα γόνατα λόγω της χαμηλής κινητικότητας των αρθρώσεων, εξ ου και η σύγκριση με το βάδισμα μιας κούκλας).
- Συμπτώματα που σχετίζονται με την ψυχική και συναισθηματική ανάπτυξη:
- σοβαρή νοητική υστέρηση,
- υπερβολικά συναισθηματική, θορυβώδης, ιδιότροπη συμπεριφορά,
- συχνό χειροκρότημα,
- εξέφραζε φιλικότητα, που τονιζόταν από ένα συνεχές χαμόγελο στο πρόσωπο,
- συχνό γέλιο χωρίς λόγο.
- Νευρολογικά συμπτώματα:
- τρόμος των άκρων,
- ανεπαρκής συντονισμός των κινήσεων με απώλεια ισορροπίας,
- μειωμένος μυϊκός τόνος,
- διάφορες διαταραχές ύπνου,
- συχνές υστερικές κρίσεις στην παιδική ηλικία,
- διαταραχές ομιλίας (το παιδί αρχίζει να μιλάει αργά, έχει κακές επικοινωνιακές δεξιότητες και ασαφή ομιλία),
- υπερκινητικότητα στο φόντο της αυξημένης διέγερσης,
- δυσκολίες στη συγκέντρωση και τη μάθηση.
Αλλά αυτή είναι μια γενικευμένη εικόνα της νόσου. Στην πραγματικότητα, η κλινική εικόνα του συνδρόμου Angelman εξαρτάται σε μεγάλο βαθμό από το στάδιο ανάπτυξης της νόσου και τον τύπο της χρωμοσωμικής μετάλλαξης που προκάλεσε την παθολογία. Αυτό σημαίνει ότι τα συμπτώματα της νόσου μπορεί να διαφέρουν σημαντικά σε διαφορετικούς ασθενείς, κάτι που για μεγάλο χρονικό διάστημα δεν μας επέτρεπε να διακρίνουμε την παθολογία από άλλες με παρόμοια κλινική εικόνα.
Μεταξύ του συνολικού αριθμού συμπτωμάτων, μπορούμε να επισημάνουμε αυτά που είναι χαρακτηριστικά όλων των ασθενών χωρίς εξαίρεση:
- σοβαρή νοητική υστέρηση,
- ακατάλληλη συμπεριφορά (παράλογο γέλιο, αυξημένη διέγερση, μειωμένη συγκέντρωση, κατάσταση ευφορίας),
- υποανάπτυξη κινητικών δεξιοτήτων,
- κακός συντονισμός των κινήσεων, αταξία βάδισης (ανώμαλος ρυθμός, ταλάντωση από πλευρά σε πλευρά, κ.λπ.), τρόμος των άκρων.
- διαταραχή ανάπτυξης λόγου με κυριαρχία μη λεκτικών μέσων επικοινωνίας.
Μεταξύ των συμπτωμάτων που αντιμετωπίζει η συντριπτική πλειοψηφία των ασθενών, μπορούν να διακριθούν τα ακόλουθα:
- δυσαναλογία μεταξύ κεφαλής και σώματος που προκαλείται από καθυστερημένη σωματική ανάπτυξη,
- σε πολλούς ασθενείς το σχήμα του κρανίου είναι τέτοιο που το μέγεθος του εγκεφάλου παραμένει μικρότερο από ό,τι σε υγιείς ανθρώπους (μικροκεφαλία),
- επιληπτικές κρίσεις πριν από την ηλικία των 3 ετών με προοδευτική μείωση της έντασης και της συχνότητας σε μεγαλύτερη ηλικία,
- παραμόρφωση των παραμέτρων του ΗΕΓ (διακυμάνσεις και υψηλό πλάτος κυμάτων χαμηλής συχνότητας).
Αυτά τα συμπτώματα είναι αρκετά συχνά, ωστόσο, το 20% των ασθενών με σύνδρομο Angelman δεν τα έχουν.
Ακόμα λιγότερο συχνά, είναι δυνατή η διάγνωση τέτοιων εκδηλώσεων της νόσου όπως:
- σοβαρός ή ήπιος στραβισμός,
- κακός έλεγχος της κίνησης της γλώσσας, με αποτέλεσμα οι ασθενείς συχνά να βγάζουν έξω τη γλώσσα τους χωρίς λόγο,
- δυσκολίες στην κατάποση και το πιπίλισμα, ειδικά σε μικρά παιδιά,
- διαταραχή της χρώσης του δέρματος και των ματιών,
- τα χέρια σηκωμένα ή λυγισμένα κατά το περπάτημα,
- υπερρεφλεξία,
- διαταραχές ύπνου, ειδικά στην παιδική ηλικία,
- συχνή σιελόρροια,
- ακόρεστη δίψα,
- υπερβολικά ενεργές κινήσεις μάσησης,
- υπερευαισθησία στη θερμότητα,
- επίπεδο πίσω μέρος του κεφαλιού,
- προεξέχουσα κάτω γνάθος,
- λείες παλάμες.
Ένα αρκετά μεγάλο ποσοστό ασθενών έχει προβλήματα με την ούρηση, τα οποία δεν ελέγχουν επαρκώς, μειωμένες λεπτές κινητικές δεξιότητες, γεγονός που δημιουργεί δυσκολίες στην αυτοφροντίδα και τη μάθηση, και υπερβολικό βάρος. Σχεδόν όλοι οι ασθενείς βιώνουν την εφηβεία αργότερα από τους υγιείς συνομηλίκους τους.
Τα παιδιά με σύνδρομο Angelman αντιλαμβάνονται καλά την προφορική ομιλία και την κατανοούν, αλλά δεν θέλουν να συμμετέχουν σε συζητήσεις, περιορίζοντας την ομιλία τους σε αρκετές δεκάδες λέξεις που είναι απαραίτητες στην καθημερινή ζωή. Ωστόσο, στην ενήλικη ζωή, αυτοί οι ασθενείς φαίνονται νεότεροι από τους συνομηλίκους τους χωρίς γενετικές παθολογίες.
Πολλά συμπτώματα του συνδρόμου Angelman είναι ασταθή, επομένως η κλινική εικόνα της νόσου αλλάζει σημαντικά με την ηλικία. Οι σπασμοί και οι επιληπτικές κρίσεις γίνονται λιγότερο συχνές ή εξαφανίζονται εντελώς, ο ασθενής γίνεται λιγότερο ευερέθιστος και ο ύπνος βελτιώνεται.
Επιπλοκές και συνέπειες
Το σύνδρομο Angelman είναι μια σοβαρή, προς το παρόν σχεδόν ανίατη χρωμοσωμική παθολογία που στερεί από τους ασθενείς την ευκαιρία να ζήσουν μια φυσιολογική ζωή. Η ζωή ενός παιδιού με σύνδρομο Angelman εξαρτάται σε μεγάλο βαθμό από τον τύπο της χρωμοσωμικής ανωμαλίας.
Ο διπλασιασμός ενός τμήματος χρωμοσώματος είναι ασύμβατος με τη ζωή στις περισσότερες περιπτώσεις. Και ακόμα κι αν τέτοιοι ασθενείς δεν πεθάνουν στη βρεφική ηλικία και φτάσουν στην εφηβεία, δεν έχουν καμία πιθανότητα να αποκτήσουν παιδιά.
Η διαγραφή ή η απουσία ενός μέρους των γονιδίων που εμφανίζεται συχνότερα στο σύνδρομο Angelman αποτελεί εμπόδιο για το παιδί να μάθει να περπατάει και να μιλάει. Τέτοια παιδιά έχουν μια πιο σοβαρή μορφή νοητικής καθυστέρησης και οι επιληπτικές κρίσεις εμφανίζονται συχνότερα και η έντασή τους είναι πολύ μεγαλύτερη από ό,τι σε ασθενείς με άλλες χρωμοσωμικές ανωμαλίες.
Εάν υπάρχει μόνο μια μετάλλαξη ενός γονιδίου, με την δέουσα προσοχή και προσέγγιση, το παιδί μπορεί να διδαχθεί τα βασικά της αυτοφροντίδας, της επικοινωνίας και της αλληλεπίδρασης σε μια ομάδα, αν και θα εξακολουθεί να υστερεί σε ανάπτυξη από τους συνομηλίκους του.
Για τα παιδιά με σύνδρομο Angelman, που είναι ευγενικά από τη φύση τους, το πιο σημαντικό είναι η αγάπη και η προσοχή των γονιών τους. Μόνο σε αυτήν την περίπτωση η εκπαίδευση του παιδιού θα αποδώσει καρπούς, έστω και μικρούς. Φυσικά, οι ασθενείς με σύνδρομο Angelman δεν θα μπορούν να φοιτήσουν σε κανονικό σχολείο. Χρειάζονται ειδικές τάξεις όπου τα παιδιά θα διδάσκονται πρώτα να συγκεντρώνονται και στη συνέχεια σταδιακά θα τους δίνονται οι βασικές σχολικές γνώσεις.
Διαγνωστικά Σύνδρομο Angelman
Το σύνδρομο Angelman είναι μια συγγενής αναπτυξιακή παθολογία. Ωστόσο, λόγω ορισμένων περιστάσεων, είναι συχνά αδύνατο να διαγνωστεί στη βρεφική ηλικία και την πρώιμη παιδική ηλικία. Αυτό οφείλεται στη μη εξειδίκευση και την ασθενή έκφραση των συμπτωμάτων σε βρέφη και παιδιά κάτω των 3 ετών. Και η επικράτηση της νόσου στη χώρα μας δεν είναι τόσο μεγάλη που οι γιατροί έχουν μάθει να την αναγνωρίζουν μεταξύ των συνομηλίκων της.
Το σύνδρομο Angelman στα βρέφη μπορεί να εκδηλωθεί ως μειωμένος μυϊκός τόνος, ο οποίος εκδηλώνεται με προβλήματα στη σίτιση (αδυναμία του αντανακλαστικού θηλασμού και κατάποσης) και αργότερα με δυσκολίες στην εκμάθηση του περπατήματος (τέτοια παιδιά αρχίζουν να περπατούν πολύ αργότερα). Αυτά τα συμπτώματα είναι τα πρώτα σημάδια μιας αναπτυξιακής ανωμαλίας στο μωρό, η οποία μπορεί κάλλιστα να σχετίζεται με μια χρωμοσωμική ανωμαλία. Μόνο η γενετική ανάλυση μπορεί να επιβεβαιώσει αυτή την υπόθεση.
Ιδιαίτερη προσοχή δίνεται στα παιδιά των οποίων οι γονείς έχουν διάφορες γονιδιωματικές ή χρωμοσωμικές διαταραχές. Άλλωστε, η ασθένεια μπορεί να μην εκδηλωθεί στην αρχή και, εάν η παθολογία εντοπιστεί εγκαίρως, ξεκινώντας εντατική εργασία με το παιδί, είναι δυνατόν να επιτευχθεί σημαντικά μεγαλύτερη επιτυχία στη μάθηση, επιβραδύνοντας την εξέλιξη της νόσου.
Εάν οι γονείς έχουν διάφορες χρωμοσωμικές ανωμαλίες, η γενετική ανάλυση πραγματοποιείται ακόμη και πριν γεννηθεί το μωρό, καθώς η SA είναι μία από τις παθολογίες που μπορούν να ανιχνευθούν στο εμβρυϊκό στάδιο.
Η συλλογή υλικού για γενετική έρευνα μπορεί να πραγματοποιηθεί με δύο τρόπους:
- επεμβατική (με ένα ορισμένο ποσοστό κινδύνου, καθώς είναι απαραίτητο να διεισδύσει η μήτρα για να ληφθεί δείγμα αμνιακού υγρού),
- μη επεμβατική (ανάλυση του DNA του μωρού από το αίμα της μητέρας).
Στη συνέχεια, διεξάγονται οι ακόλουθες μελέτες:
- φθορίζουσα in situ υβριδοποίηση (μέθοδος FISH) – σύνδεση ενός ανιχνευτή DNA επισημασμένου με ειδική χρωστική στο υπό μελέτη DNA, ακολουθούμενη από εξέταση υπό μικροσκόπιο.
- ανάλυση μεταλλάξεων στο γονίδιο ube3a και σε αποτυπωμένα γονίδια,
- Ανάλυση μεθυλίωσης DNA χρησιμοποιώντας ειδικές μεθόδους που χρησιμοποιούνται στη γενετική.
Οι γενετικές εξετάσεις παρέχουν αρκετά ακριβείς πληροφορίες σε περίπτωση χρωμοσωμικών ανωμαλιών, πράγμα που σημαίνει ότι οι μελλοντικοί γονείς γνωρίζουν εκ των προτέρων για τι να προετοιμαστούν. Ωστόσο, υπάρχουν εξαιρέσεις. Σε μια συγκεκριμένη ομάδα ασθενών, παρουσία όλων των συμπτωμάτων που υποδηλώνουν παθολογία, τα αποτελέσματα των εξετάσεων παραμένουν φυσιολογικά. Δηλαδή, η παθολογία μπορεί να εντοπιστεί μόνο με προσεκτική παρατήρηση του παιδιού από την πρώιμη παιδική ηλικία: πώς τρώει, πότε άρχισε να περπατάει και να μιλάει, αν λυγίζει τα πόδια του όταν περπατάει κ.λπ.
Εκτός από τη μέθοδο FISH, μεταξύ των οργανικών διαγνωστικών μεθόδων για το σύνδρομο Angelman, μπορεί κανείς να ξεχωρίσει την τομογραφία (CT ή MRI), η οποία βοηθά στον προσδιορισμό της κατάστασης και του μεγέθους του εγκεφάλου, και ένα ηλεκτροεγκεφαλογράφημα (EEG), το οποίο δείχνει πώς λειτουργούν τα μεμονωμένα μέρη του εγκεφάλου.
Οι γιατροί συνήθως κάνουν μια τελική διάγνωση στην ηλικία των 3-7 ετών, όταν ο ασθενής έχει ήδη τα περισσότερα συμπτώματα και η δυναμική της εξέλιξης της νόσου είναι ορατή.
Ποιες δοκιμές χρειάζονται;
Διαφορική διάγνωση
Το σύνδρομο Angelman είναι μια γενετική παθολογία που ουσιαστικά δεν έχει συγκεκριμένες εκδηλώσεις. Τα περισσότερα συμπτώματα μπορούν εξίσου να υποδηλώνουν τόσο το AS όσο και άλλες γενετικές παθολογίες.
Η διαφορική διάγνωση του συνδρόμου Angelman πραγματοποιείται με τις ακόλουθες παθολογίες:
- Σύνδρομο Pitt-Hopkins (οι ασθενείς χαρακτηρίζονται από νοητική υστέρηση, χαρούμενο χαρακτήρα, χαμογελαστοί, έχουν μάλλον μεγάλο και φαρδύ στόμα, παρατηρείται μικροκεφαλία). Η διαφορά είναι οι κρίσεις υπεραερισμού και η συγκράτηση της αναπνοής σε κατάσταση αφύπνισης.
- Σύνδρομο Christianson (οι ασθενείς είναι άτομα με νοητική υστέρηση με χαρούμενη διάθεση, ανίκανα να μιλήσουν, που χαρακτηρίζονται από μικροκεφαλία, αταξία, σπασμούς, ακούσιες μυϊκές κινήσεις).
- Σύνδρομο Mowat-Wilson (συμπτώματα: νοητική υστέρηση, επιληπτικές κρίσεις, μυτερό πηγούνι, ανοιχτό στόμα, χαρούμενη έκφραση στο πρόσωπο, μικροκεφαλία). Διάκριση: μεγάλη απόσταση μεταξύ των ματιών, μάτια κεκλιμένα προς τα μέσα, στρογγυλεμένη άκρη της μύτης, πτερύγιο στραμμένο προς τα πίσω.
- Σύνδρομο Kabuki (χαρακτηρίζεται από ήπια έως μέτρια νοητική υστέρηση, προβλήματα ομιλίας και κινητικότητας, μυϊκή αδυναμία, επιληπτικές κρίσεις, μικροκεφαλία, μεγάλα διαστήματα μεταξύ κνησμών και μειωμένο συντονισμό). Χαρακτηρίζεται από τοξωτά φρύδια, ανεστραμμένο πλάγιο τμήμα του κάτω βλεφάρου, ευρύχωρα μάτια, μακριές βλεφαρικές σχισμές με μακριές, πυκνές βλεφαρίδες.
- Σύνδρομο Rett (διαφοροποίηση από το AS στις γυναίκες). Συμπτώματα: καθυστέρηση στην ανάπτυξη της ομιλίας, επιληπτικές κρίσεις, μικροκεφαλία. Η διαφορά είναι ότι δεν υπάρχει χαρούμενη έκφραση στο πρόσωπο, υπάρχουν κρίσεις άπνοιας και απραξίας, η οποία εξελίσσεται με την πάροδο του χρόνου.
- Σύνδρομο αυτοσωμικής υπολειπόμενης νοητικής καθυστέρησης 38 (συμπτώματα: έντονη νοητική υστέρηση με καθυστερήσεις στις κινητικές δεξιότητες και την ομιλία, μυϊκή αδυναμία, προβλήματα σίτισης στη βρεφική ηλικία, παρορμητικότητα). Διακριτικό χαρακτηριστικό είναι το μπλε χρώμα της ίριδας.
- Σύνδρομο διπλασιασμού του γονιδίου MECP 2 (διαφοροποίηση από SA σε άνδρες). Συμπτώματα: σοβαρή νοητική υστέρηση, μυϊκή αδυναμία από την παιδική ηλικία, προβλήματα ομιλίας ή έλλειψη ομιλίας, επιληψία. Διακρίσεις: προοδευτική μυοπάθεια, συνεχώς υποτροπιάζουσες λοιμώξεις.
- Σύνδρομο Kleefstra (συμπτώματα: προβλήματα ομιλίας και σκέψης, μυϊκή αδυναμία, διαταραχές ύπνου, έλλειψη προσοχής, ανοιχτό στόμα, υπερκινητικότητα, επιληπτικές κρίσεις, αταξία, διαταραχές ισορροπίας). Διακριτικά χαρακτηριστικά: επίπεδο πρόσωπο, κοντή, στριμμένη μύτη, μάτια με μεγάλα ανοίγματα, μεγάλο, ανεστραμμένο κάτω χείλος, επιθετικά ξεσπάσματα.
- Σύνδρομο Smith-Magenis (χαρακτηρίζεται από επιληπτικές κρίσεις, προβλήματα ύπνου, διαταραχές νοητικής και κινητικής ανάπτυξης). Διακριτικά χαρακτηριστικά περιλαμβάνουν ένα πλατύ και επίπεδο πρόσωπο και ένα προεξέχον μέτωπο.
- Σύνδρομο Koolen-de Vries (ήπια έως μέτρια νοητική υστέρηση, μυϊκή αδυναμία, επιληπτικές κρίσεις, φιλικότητα). Διακριτικά χαρακτηριστικά: μακρύ πρόσωπο με ψηλό μέτωπο, προεξέχοντα αυτιά, λοξά μάτια, υψηλή κινητικότητα των αρθρώσεων, συγγενείς καρδιοπάθειες.
- Σύνδρομο Phelan-McDermid (συμπτώματα: νοητική υστέρηση, διαταραχές ομιλίας ή έλλειψη ομιλίας). Διακρίσεις: μεγάλα χέρια με ανεπτυγμένους μύες, μυϊκή αδυναμία εκ γενετής, ασθενής εφίδρωση.
Τέτοιες παθολογίες όπως η ανεπάρκεια αδενυλικού ηλεκτρικού, το αυτοσωμικό υπολειπόμενο σύνδρομο νοητικής καθυστέρησης 1, το σύνδρομο επανάληψης του χρωμοσώματος 2q23.1, τα σύνδρομα απλοανεπάρκειας γονιδίων FOXG1, STXBP1 ή MEF2C και ορισμένες άλλες μπορούν να «καυχηθούν» για συμπτώματα παρόμοια με το σύνδρομο Angelman.
Το καθήκον του γιατρού είναι να κάνει μια ακριβή διάγνωση, διαφοροποιώντας το σύνδρομο Angelman από παθολογίες με παρόμοια συμπτώματα και να συνταγογραφήσει αποτελεσματική θεραπεία που να είναι σχετική με το διαγνωσμένο στάδιο της νόσου.
Ποιος θα επικοινωνήσει;
Θεραπεία Σύνδρομο Angelman
Το σύνδρομο Angelman είναι μια από εκείνες τις παθολογίες για τις οποίες η ιατρική εξακολουθεί να αναζητά αποτελεσματική θεραπεία. Η αιτιολογική θεραπεία της νόσου βρίσκεται στο στάδιο της ανάπτυξης με διάφορες μεθόδους και μέσα, πολλά από τα οποία δεν έχουν ακόμη δοκιμαστεί σε ανθρώπους. Αυτό σημαίνει ότι προς το παρόν οι γιατροί πρέπει να περιοριστούν στη συμπτωματική θεραπεία, η οποία βοηθά με κάποιο τρόπο να ανακουφιστεί η δυσάρεστη κατάσταση των παιδιών και των ενηλίκων με σύνδρομο μαριονέτας, που πάσχουν από επιληπτικές κρίσεις, σιελόρροια, υπόταση και διαταραχές ύπνου.
Έτσι, είναι δυνατόν να μειωθεί η συχνότητα και η ένταση των επιληπτικών κρίσεων με τη βοήθεια ενός σωστά επιλεγμένου αντισπασμωδικού φαρμάκου. Αλλά όλη η δυσκολία έγκειται στο ότι οι κρίσεις σε ασθενείς με SA διαφέρουν από τις συνηθισμένες επιληπτικές κρίσεις στο ότι χαρακτηρίζονται από διάφορους τύπους κρίσεων, πράγμα που σημαίνει ότι η κατάσταση μπορεί να ανακουφιστεί με τη χορήγηση αρκετών φαρμάκων ταυτόχρονα.
Τα πιο δημοφιλή αντισπασμωδικά που χρησιμοποιούνται για τη θεραπεία του συνδρόμου Angelman είναι: βαλπροϊκό οξύ, τοπιραμάτη, λαμοτριγίνη, λεβετιρακετάμη, κλοναζεπάμη και φάρμακα που βασίζονται σε αυτά. Λιγότερο συχνά χρησιμοποιούνται φάρμακα που βασίζονται στην καρμαζεπίνη, τη φαινυτοΐνη, τη φαινοβαρβιτάλη, την εθοσουξιμίδη, καθώς μερικά από αυτά μπορούν να προκαλέσουν ένα παράδοξο αποτέλεσμα που συνίσταται στην ενίσχυση και αύξηση της συχνότητας των επιληπτικών κρίσεων. Αυτό συμβαίνει εάν το φάρμακο χρησιμοποιείται ως μέρος μονοθεραπείας.
Για την αντιμετώπιση της σιελόρροιας, χρησιμοποιούνται συνήθως δύο μέθοδοι: η φαρμακευτική (φάρμακα που καταστέλλουν την παραγωγή σάλιου) και η χειρουργική, η οποία περιλαμβάνει την επανεμφύτευση των σιελογόνων πόρων. Αλλά στην περίπτωση της σαλιγκαροπάθειας, αυτές οι μέθοδοι θεωρούνται αναποτελεσματικές και το ζήτημα παραμένει ανοιχτό. Οι γονείς και όσοι φροντίζουν τέτοιους ασθενείς πρέπει να δώσουν ιδιαίτερη προσοχή σε αυτό το ζήτημα, καθώς οι ίδιοι οι ασθενείς συνήθως δεν ελέγχουν την σιελόρροια και κάποιοι απλώς δεν μπορούν να φροντίσουν τον εαυτό τους.
Ένα άλλο πρόβλημα είναι η μικρή διάρκεια ύπνου. Συχνά τα παιδιά με σύνδρομο Angelman κοιμούνται όχι περισσότερο από 5 ώρες, κάτι που έχει αρνητικό αντίκτυπο στη λειτουργία ολόκληρου του σώματος. Τα παιδιά που ευερεθίζονται εύκολα, είναι δραστήρια και αγαπούν τα παιχνίδια και την επικοινωνία (ακόμα κι αν προσπαθούν να περιοριστούν σε μη λεκτικές μεθόδους) είναι αισθητά κουρασμένα κατά τη διάρκεια της ημέρας. Για να ξεκουραστεί καλά, το σώμα χρειάζεται έναν βαθύ, πλήρη ύπνο, αλλά αυτό ακριβώς είναι το πρόβλημα.
Φαίνεται ότι τα ηρεμιστικά φάρμακα (φαινοθειαζίνες και άτυπα αντιψυχωσικά) που ηρεμούν το νευρικό σύστημα θα πρέπει να επαρκούν για τη βελτίωση του ύπνου σε ευερέθιστους ασθενείς. Αλλά στην περίπτωση της ΑΣ, η χρήση τέτοιων φαρμάκων είναι γεμάτη με την εμφάνιση αρνητικών επιπτώσεων. Επομένως, οι γιατροί εξακολουθούν να προτιμούν ήπια υπνωτικά χάπια, όπως η μελατονίνη (ένα φυσικό ορμονικό φάρμακο που βασίζεται στην ορμόνη του ύπνου), η οποία χορηγείται στους ασθενείς μία ώρα πριν τον ύπνο σε ποσότητα 1 δισκίου, και η διφαινυδραμίνη. Η συχνότητα χορήγησης και η δοσολογία της οποίας καθορίζονται από τον γιατρό ανάλογα με την κατάσταση και την ηλικία του ασθενούς.
Μερικές φορές οι ασθενείς με σύνδρομο Angelman έχουν προβλήματα με την πέψη και τα κόπρανα. Μπορείτε να βελτιώσετε τα κόπρανά σας με καθαρτικά (κατά προτίμηση φυτικά).
Ή μπορείτε να προσεγγίσετε το πρόβλημα διαφορετικά, όπως έκαναν οι Αμερικανοί γιατροί, βασιζόμενοι σε ορισμένες μεθόδους θεραπείας του αυτισμού, επειδή πολλά συμπτώματα που χαρακτηρίζουν το AS είναι επίσης χαρακτηριστικά του αυτισμού (παρορμητικότητα, ακούσιες κινήσεις, επαναλαμβανόμενες ενέργειες, έλλειψη προσοχής, προβλήματα επικοινωνίας κ.λπ.). Σημειώθηκε ότι η εισαγωγή της ορμόνης σεκρετίνης, η οποία ομαλοποιεί την πέψη και τα κόπρανα, έχει θετική επίδραση στην προσοχή των ασθενών και η οξυτοκίνη βοηθά στη βελτίωση των γνωστικών ικανοτήτων και της μνήμης του παιδιού, καθώς και στη σωστή συμπεριφορά.
Είναι αλήθεια ότι οι ορμόνες από μόνες τους δεν αρκούν, ειδικά όταν πρόκειται για παιδιά. Στο σύνδρομο Angelman, ενδείκνυται η συμπεριφορική θεραπεία, η συνεργασία με ψυχολόγο και λογοθεραπευτή (διδασκαλία μεθόδων μη λεκτικής επικοινωνίας και νοηματικής γλώσσας). Η εκπαίδευση τέτοιων παιδιών θα πρέπει να βασίζεται σε ένα ατομικό πρόγραμμα με τη συμμετοχή ειδικά εκπαιδευμένων εκπαιδευτικών, ψυχολόγου και γονέων. Δυστυχώς, αυτό δεν είναι εφικτό παντού και οι οικογένειες μένουν μόνες με το πρόβλημά τους.
Δεδομένου ότι πολλοί νεαροί ασθενείς με ΑΣ υποφέρουν από χαμηλό μυϊκό τόνο και προβλήματα στις αρθρώσεις, δίνεται μεγάλη προσοχή στη φυσικοθεραπεία. Τις περισσότερες φορές, οι γιατροί καταφεύγουν στη χρήση εφαρμογών παραφίνης, ηλεκτροφόρησης και μαγνητικής θεραπείας.
Το ενεργητικό τονωτικό μασάζ και οι ειδικές ασκήσεις θεραπευτικής σωματικής άσκησης θα βοηθήσουν το άρρωστο παιδί να σταθεί στα πόδια του και να περπατήσει με αυτοπεποίθηση μετά από λίγο. Η γυμναστική στο νερό είναι ιδιαίτερα χρήσιμη από αυτή την άποψη, η οποία συνιστάται για γυμναστική σε δροσερό νερό. Αυξάνει τον μυϊκό τόνο και διδάσκει στο παιδί να ελέγχει το σώμα του και να συντονίζει τις κινήσεις.
Αντισπασμωδική θεραπεία
Το πιο επικίνδυνο σύμπτωμα του συνδρόμου Angelman είναι οι κρίσεις παρόμοιες με αυτές της επιληψίας. Αυτό το σύμπτωμα παρατηρείται στο 80% των ασθενών, πράγμα που σημαίνει ότι σε όλους τους ασθενείς πρέπει να συνταγογραφείται αποτελεσματική αντισπασμωδική αγωγή.
Η θεραπεία των επιληπτικών κρίσεων πραγματοποιείται με τη βοήθεια βιταμινών και αντισπασμωδικών. Στο σύνδρομο Angelman, που συνοδεύεται από σπασμωδικό σύνδρομο, οι βιταμίνες της ομάδας Β, καθώς και οι βιταμίνες C, D και E θα είναι χρήσιμες. Αλλά η συνταγογράφηση βιταμινοθεραπείας μόνοι σας σε αυτή την περίπτωση είναι πολύ επικίνδυνη, επειδή η ανεξέλεγκτη λήψη βιταμινών μπορεί να μειώσει την αποτελεσματικότητα των αντιεπιληπτικών φαρμάκων και να προκαλέσει νέες, πιο σοβαρές και παρατεταμένες κρίσεις.
Η επιλογή των αντισπασμωδικών φαρμάκων και η συνταγογράφηση της αποτελεσματικής δοσολογίας τους θα πρέπει επίσης να γίνεται από εξειδικευμένο γιατρό. Αυτός ή αυτή αποφασίζει επίσης εάν ένα φάρμακο θα είναι αρκετό ή εάν ο ασθενής θα πρέπει να λαμβάνει 2 ή περισσότερα φάρμακα για μεγάλο χρονικό διάστημα.
Για τους περισσότερους ασθενείς, οι γιατροί συνταγογραφούν φάρμακα βαλπροϊκού οξέος (Valproic acid, Depakine, Convulex, Valparin, κ.λπ.), τα οποία προλαμβάνουν τις επιληπτικές κρίσεις και βελτιώνουν τη διάθεση και την ψυχική κατάσταση των ασθενών.
Το βαλπροϊκό οξύ διατίθεται με τη μορφή δισκίων, σιροπιού και ενέσιμων διαλυμάτων. Το πιο δημοφιλές φάρμακο είναι το φάρμακο παρατεταμένης αποδέσμευσης "Depakine" σε δισκία και ως διάλυμα για ενδοφλέβια χορήγηση. Η δοσολογία του φαρμάκου καθορίζεται από τον γιατρό ξεχωριστά, ανάλογα με το βάρος, την ηλικία και την κατάσταση του ασθενούς.
Το φάρμακο λαμβάνεται κατά τη διάρκεια των γευμάτων 2 έως 3 φορές την ημέρα. Η μέση ημερήσια δόση είναι 20-30 mg ανά 1 κιλό βάρους του ασθενούς, η μέγιστη είναι 50 mg/kg την ημέρα.
Αντενδείξεις χρήσης. Μην το χρησιμοποιείτε σε περίπτωση δυσλειτουργίας του ήπατος και του παγκρέατος, αιμορραγικής διάθεσης, ηπατίτιδας, πορφυρίας και υπερευαισθησίας στο φάρμακο.
Οι παρενέργειες περιλαμβάνουν τρέμουλο στα χέρια, διαταραχές του πεπτικού συστήματος και των κοπράνων, καθώς και αλλαγές στο σωματικό βάρος.
Η «τοπιραμάτη» είναι επίσης φάρμακο επιλογής για τη ΣΑ. Παράγεται σε μορφή δισκίου και χρησιμοποιείται τόσο ως μέρος μονοθεραπείας όσο και σε συνδυασμό με άλλα φάρμακα.
Τρόπος χορήγησης και δοσολογία. Λαμβάνετε τα δισκία από το στόμα ανεξάρτητα από την πρόσληψη τροφής. Η αρχική ημερήσια δόση για ενήλικες είναι 25-50 mg, για παιδιά - 0,5-1 mg/kg. Κάθε εβδομάδα, η δόση αυξάνεται σύμφωνα με τις οδηγίες του γιατρού.
Το φάρμακο δεν πρέπει να λαμβάνεται κατά τη διάρκεια της εγκυμοσύνης και της γαλουχίας, καθώς και σε περίπτωση υπερευαισθησίας στα συστατικά του. Το φάρμακο έχει πολλές διαφορετικές παρενέργειες.
Φάρμακα που μπορεί να συνταγογραφήσει ένας γιατρός για το σύνδρομο Angelman: Κλομαζεπάμη, Ριβοτρίλη, Λαμοτριγίνη, Σεϊζάρ, Λαμικτάλ, Λεβετιρακετάμη, Κέπρα, Επιτέρα, κ.λπ.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Παραδοσιακή ιατρική και ομοιοπαθητική
Η παραδοσιακή ιατρική, όπως και τα ομοιοπαθητικά σκευάσματα, είναι φυσικά σχετικά ασφαλή, αλλά η αποτελεσματικότητα μιας τέτοιας θεραπείας για το σύνδρομο Angelman μπορεί να θεωρηθεί αμφιλεγόμενη.
Αν και η λαϊκή θεραπεία μπορεί να βοηθήσει σε ορισμένα πράγματα. Μιλάμε για την παύση των επιληπτικών κρίσεων. Από αυτή την άποψη, η φυτική θεραπεία μπορεί να είναι αρκετά αποτελεσματική.
Ένα καλό αποτέλεσμα παρέχεται από μια φαρμακευτική συλλογή με βάση την παιώνια, τη γλυκόριζα και το φύκι (τα συστατικά λαμβάνονται σε ίσες ποσότητες). Τα βότανα πρέπει να αλέθονται σε αλεύρι. Μετά από 2 εβδομάδες από την έναρξη της λήψης, μπορείτε να παρατηρήσετε μια σημαντική μείωση στη συχνότητα των επιληπτικών κρίσεων.
Το αφέψημα λεβάντας (1 κουταλάκι του γλυκού ανά ποτήρι βραστό νερό) είναι επίσης χρήσιμο για κράμπες. Το μείγμα βράζεται για 5 λεπτά και εγχύεται για μισή ώρα. Το φάρμακο λαμβάνεται το βράδυ για 14 ημέρες.
Ένα υδατικό (ή αλκοολούχο) έγχυμα από μηδικό θεωρείται αποτελεσματικό για επιληπτικές κρίσεις.
Από τα ομοιοπαθητικά φάρμακα για την πρόληψη των επιληπτικών κρίσεων στο σύνδρομο Angelman, μπορείτε να χρησιμοποιήσετε φάρμακα με βάση το χαμομήλι και το motherwort, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Αλλά πρέπει να ληφθεί υπόψη ότι μόνο ένας ομοιοπαθητικός γιατρός μπορεί να συνταγογραφήσει αποτελεσματικές και ασφαλείς δόσεις φαρμάκων σε κάθε συγκεκριμένη περίπτωση.
Πρόληψη
Όπως πιθανώς έχει ήδη καταλάβει ο αναγνώστης, η ιατρική δεν είναι ακόμη σε θέση να αποτρέψει τις γονιδιακές μεταλλάξεις και άλλες χρωμοσωμικές ανωμαλίες, ωστόσο, καθώς και να διορθώσει την κατάσταση. Αυτό μπορεί να συμβεί σε οποιονδήποτε, επειδή τα παιδιά με σύνδρομο Angelman γεννιούνται από υγιείς γονείς και η γενετική, η οποία είναι σήμερα ένας από τους λιγότερο μελετημένους κλάδους της ιατρικής, δεν μπορεί ακόμη να το εξηγήσει αυτό.
Το μόνο που μπορεί να γίνει είναι να υιοθετηθεί μια υπεύθυνη προσέγγιση στον προγραμματισμό της εγκυμοσύνης, να εγγραφεί και να υποβληθεί σε εξετάσεις εγκαίρως. Αλλά και πάλι, ένα τέτοιο μέτρο θα είναι περισσότερο εκπαιδευτικό παρά προληπτικό, όπως οποιεσδήποτε εξετάσεις. Αλλά οι νέοι γονείς θα γνωρίζουν εκ των προτέρων για τι να προετοιμαστούν και, σε περίπτωση θετικής απάντησης, θα αποφασίσουν οι ίδιοι αν μπορούν να αναλάβουν την ευθύνη της ανατροφής ενός άρρωστου παιδιού.
Πρόβλεψη
Η πρόγνωση για το σύνδρομο Angelman εξαρτάται από τη φύση της χρωμοσωμικής ανωμαλίας και την έγκαιρη ανίχνευσή της. Τα παιδιά που πλήττονται περισσότερο είναι εκείνα των οποίων το χρωμόσωμα 15 περιέχει «κενά» σε γονίδια (διαγραφή). Η πιθανότητα τέτοιοι ασθενείς να περπατούν και να μιλάνε είναι εξαιρετικά χαμηλή. Άλλες περιπτώσεις μπορούν να διορθωθούν με προσεκτική προσέγγιση και αγάπη για το παιδί σας.
Δυστυχώς, τέτοιοι ασθενείς δεν θα μπορέσουν να γίνουν πλήρη μέλη της κοινωνίας, παρά το γεγονός ότι δεν είναι καθόλου ηλίθιοι, κατανοούν την ομιλία και το νόημά της. Ωστόσο, θα έχουν προβλήματα με την επικοινωνία για το υπόλοιπο της ζωής τους. Οι ασθενείς μπορούν να διδαχθούν νοηματική γλώσσα από την παιδική ηλικία, αλλά δεν μπορούν να αναγκαστούν να επικοινωνούν χρησιμοποιώντας λέξεις. Το λεξιλόγιο των «ομιλούντων» ασθενών περιορίζεται στο ελάχιστο των λέξεων που χρησιμοποιούνται στην καθημερινή ζωή (5-15 λέξεις).
Όσον αφορά το προσδόκιμο ζωής και τη γενική υγεία των ασθενών με σύνδρομο Angelman, τα στοιχεία εδώ κυμαίνονται γύρω από τις μέσες τιμές. Στην ενήλικη ζωή, οι ασθενείς αντιμετωπίζουν κυρίως προβλήματα υγείας όπως η σκολίωση και η παχυσαρκία, τα οποία, με τη σωστή προσέγγιση στη θεραπεία, δεν απειλούν τη ζωή.