Ιατρικός εμπειρογνώμονας του άρθρου

Νέες δημοσιεύσεις
Η αλκαπτονουρία είναι μια συγγενής ενζυμική ανωμαλία
Τελευταία επισκόπηση: 04.07.2025

Όλα τα περιεχόμενα του iLive ελέγχονται ιατρικά ή ελέγχονται για να διασφαλιστεί η όσο το δυνατόν ακριβέστερη ακρίβεια.
Έχουμε αυστηρές κατευθυντήριες γραμμές προμήθειας και συνδέουμε μόνο με αξιόπιστους δικτυακούς τόπους πολυμέσων, ακαδημαϊκά ερευνητικά ιδρύματα και, όπου είναι δυνατόν, ιατρικά επισκοπικά μελέτες. Σημειώστε ότι οι αριθμοί στις παρενθέσεις ([1], [2], κλπ.) Είναι σύνδεσμοι με τις οποίες μπορείτε να κάνετε κλικ σε αυτές τις μελέτες.
Εάν πιστεύετε ότι κάποιο από το περιεχόμενό μας είναι ανακριβές, παρωχημένο ή αμφισβητήσιμο, παρακαλώ επιλέξτε το και πατήστε Ctrl + Enter.
Μία από τις πολύ σπάνιες μεταβολικές διαταραχές, η αλκαπτονουρία, αναφέρεται σε συγγενείς ανωμαλίες στον μεταβολισμό του αμινοξέος τυροσίνη.
Αυτό το σύνδρομο μπορεί επίσης να ονομάζεται ανεπάρκεια ομογενοποιητικής οξειδάσης, ομογεντισινουρία, κληρονομική ωχρόνωση ή νόσος των μαύρων ούρων.[ 1 ]
Επιδημιολογία
Σύμφωνα με στατιστικά στοιχεία, δεν υπάρχουν περισσότερες από εννέα περιπτώσεις αλκαπτονουρίας ανά 1 εκατομμύριο ανθρώπους. Και στις περισσότερες ευρωπαϊκές χώρες, υπάρχει μία περίπτωση ανά 100-250 χιλιάδες γεννήσεις.
Μεταξύ των ευρωπαϊκών χωρών, εξαίρεση αποτελεί η Σλοβακία (ειδικά η σχετικά μικρή βορειοδυτική περιοχή), όπου η συχνότητα εμφάνισης αλκαπτονουρίας είναι μία περίπτωση ανά 19.000 νεογνά. Αυτό πιθανότατα οφείλεται στο γεγονός ότι μεταξύ των οικογενειών Σλοβάκων Ρομά που ζουν εκεί, το επίπεδο ενδογαμίας (γάμος μεταξύ ξαδέρφων) είναι το υψηλότερο στην Ευρώπη: 10-14%. [ 2 ]
Αιτίες αλκαπτονουρία
Οι ακριβείς αιτίες της αλκαπτονουρίας, ως συγγενούς διαταραχής του καταβολισμού (μεταβολικής διάσπασης) του αρωματικού (ομοκυκλικού) α-αμινοξέος τυροσίνης, έχουν διαπιστωθεί: αυτός ο τύπος μεταβολικής διαταραχής είναι συνέπεια ομόζυγων ή σύνθετων ετερόζυγων μεταλλάξεων ενός από τα χιλιάδες γονίδια στο χρωμόσωμα 3, πιο συγκεκριμένα, του γονιδίου HGD στη θέση 3q21-q23 στον μακρύ βραχίονα του χρωμοσώματος. Αυτό το γονίδιο κωδικοποιεί τις νουκλεοτιδικές αλληλουχίες του ηπατικού ενζύμου ομογεντισικό-1,2-διοξυγενάση [ 3 ] (που ονομάζεται επίσης ομογεντισικό οξύ οξειδάση ή ομογεντισικό οξύ οξειδάση) - μιας μεταλλοπρωτεΐνης που περιέχει σίδηρο και είναι απαραίτητη για ένα από τα στάδια της διάσπασης της τυροσίνης στο σώμα. [ 4 ], [ 5 ]
Έτσι, η αλκαπτονουρία είναι ένα ελάττωμα του ενζύμου ομογενοποιητικό-1,2-διοξυγενάση, ή, ακριβέστερα, το αποτέλεσμα της γενετικά καθορισμένης ανεπάρκειας ή πλήρους απουσίας του. [ 6 ]
Όντας μια συγγενής ενζυμική ανεπάρκεια, η αλκαπτονουρία κληρονομείται ως αυτοσωμικό υπολειπόμενο χαρακτηριστικό, δηλαδή, για να εμφανιστεί αλκαπτονουρία στα παιδιά, και οι δύο γονείς πρέπει να έχουν ένα τροποποιημένο γονίδιο για το ένζυμο, καθώς ο καθένας από τους δύο μεταβιβάζει στο παιδί μόνο ένα αντίγραφο του γονιδίου από τα δύο διαθέσιμα.
Σύμφωνα με τα τελευταία δεδομένα, υπάρχουν περισσότερες από διακόσιες παραλλαγές τροποποίησης του γονιδίου HGD, και συχνότερα παρατηρούνται μεταλλάξεις missense, μετατόπιση και μάτισμα.
Παράγοντες κινδύνου
Ο μόνος παράγοντας κινδύνου για την ανάπτυξη αυτής της συγγενούς ενζυμοπάθειας είναι η παρουσία της στο οικογενειακό ιστορικό και η κληρονομικότητα δύο τροποποιημένων αντιγράφων του γονιδίου HGD, εάν οι γονείς δεν εμφανίζουν αλκαπτονουρία (ο κίνδυνος μετάδοσης της ανωμαλίας είναι 25%) ή εάν ένας από τους γονείς έχει αυτή τη διαταραχή. [ 7 ]
Παθογένεση
Η τυροσίνη παίζει βασικό ρόλο στη σύνθεση πρωτεϊνών, στην παραγωγή χρωμοπρωτεϊνών - της χρωστικής του δέρματος μελανίνης, καθώς και θυρεοειδικών ορμονών και νευροδιαβιβαστών κατεχολαμινών.
Ο μηχανισμός ρύθμισης της ποσότητας τυροσίνης στα κύτταρα είναι πολύπλοκος και το σώμα ομαλοποιεί την περίσσεια περιεκτικότητάς της διασπώντας την. Η διαδικασία του καταβολισμού της τυροσίνης, όπως όλα τα αρωματικά αμινοξέα, είναι πολυσταδιακή και λαμβάνει χώρα σε διάφορα στάδια. Κάθε στάδιο της μεταβολικής διάσπασης της τυροσίνης συμβαίνει με τη συμμετοχή ενός συγκεκριμένου ενζύμου και τον σχηματισμό μιας ενδιάμεσης ένωσης.
Έτσι, πρώτα το αμινοξύ διασπάται σε παρα-υδροξυφαινυλοπυροσταφυλικό, το οποίο μετατρέπεται σε αλκαπτόνη – 2,5-διυδροξυφαινυλοξικό ή ομογεντισικικό οξύ. Στη συνέχεια, η αλκαπτόνη θα πρέπει να μετατραπεί σε μαλεοξικό οξύ, αλλά αυτό δεν συμβαίνει. [ 8 ]
Και η παθογένεση της αλκαπτονουρίας συνίσταται στην παύση των βιοχημικών αντιδράσεων του καταβολισμού της τυροσίνης στο στάδιο του σχηματισμού του ομογενοποιητικού οξέος: απλά δεν υπάρχει κανένα ένζυμο που να χρειάζεται για να το διασπάσει - η οξειδάση του ομογενοποιητικού οξέος.
Το ομογεντισινικό οξύ δεν χρησιμοποιείται από τον οργανισμό και μπορεί να συσσωρευτεί με την απέκκριση μέσω των νεφρών. Επιπλέον, οξειδώνεται σε βενζοκινοοξικό (βενζοκινονοξικό οξύ), το οποίο, συνδεόμενο με τα μόρια των ιστών και των σωματικών υγρών, σχηματίζει βιοπολυμερείς ενώσεις με χρώμα που μοιάζει με μελανίνη.
Η συσσώρευση αυτών των ενδιάμεσων προϊόντων στον ιστό οδηγεί σε διαταραχή της δομής του κολλαγόνου του χόνδρινου ιστού, η οποία μειώνει την ελαστικότητά του – με την εμφάνιση πολλών κλινικών συμπτωμάτων αλκαπτονουρίας και την ανάπτυξη επιπλοκών.
Συμπτώματα αλκαπτονουρία
Η αλκαπτονουρία στα νεογνά και τα βρέφη χαρακτηρίζεται από σκουρόχρωμα ούρα. Όταν εκτίθενται στον αέρα, τα ούρα στις πάνες, τα εσώρουχα και τα εσώρουχα γίνονται σκούρα καφέ. Αυτό οφείλεται στη συσσώρευση και απελευθέρωση ομογεντισινικού οξέος, το οποίο οξειδώνεται σε βενζοκινοοξικό άλας. [ 9 ]
Ελλείψει άλλων συμπτωμάτων, η αλκαπτονουρία σε μικρά παιδιά συχνά δεν αναγνωρίζεται έγκαιρα, καθώς τα ούρα μπορεί να σκουρύνουν μετά από αρκετές ώρες ούρησης. Σύμφωνα με ορισμένα στοιχεία, μόνο το ένα πέμπτο των παιδιών κάτω των 12 μηνών που γεννήθηκαν με αυτή την ανεπάρκεια ενζύμου εντοπίζονται σε κλινικό περιβάλλον. Επομένως, είναι πολύ σημαντικό οι γονείς να δίνουν προσοχή στη φροντίδα των βρεφών τους.
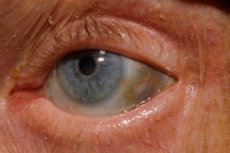
Επιπλέον, τα πρώιμα σημάδια περιλαμβάνουν μελάγχρωση (γαλαζωπό-γκρι χρώμα) του σκληρού χιτώνα των ματιών και των χόνδρων των αυτιών και της μύτης, η οποία συχνά ονομάζεται ωχρόνωση.[ 10 ]
Με την πάροδο του χρόνου, εμφανίζονται και άλλα συμπτώματα:
- έντονη χρώση του δέρματος στα ζυγωματικά, τις μασχάλες και τα γεννητικά όργανα.
- λεκέδες ρούχων όταν έρχονται σε επαφή με ιδρωμένες περιοχές του σώματος.
- κρίσεις γενικής αδυναμίας.
- βραχνή φωνή.
Θα πρέπει να ληφθεί υπόψη ότι η αλκαπτονουρία και η ωχρόνωση, όπως σημειώθηκε παραπάνω, είναι συνώνυμα ονομάτων για την ίδια διαταραχή του καταβολισμού της τυροσίνης.
Νόσος ούρων από σιρόπι σφενδάμου και αλκαπτονουρία. Η συγγενής νόσος ούρων από σιρόπι σφενδάμου ή λευκίνωση είναι επίσης μια μεταβολική διαταραχή, έχει το ίδιο πρότυπο κληρονομικότητας και ακόμη και μεταλλάξεις εμφανίζονται στο ίδιο χρωμόσωμα, αλλά επηρεάζουν το γονίδιο που κωδικοποιεί το σύμπλεγμα ενζύμων της διακλαδισμένης αλυσίδας α-κετο όξινης αφυδρογονάσης. Εξαιτίας αυτού, το σώμα δεν μπορεί να διασπάσει ορισμένα συστατικά των πρωτεϊνών, ιδιαίτερα τα αμινοξέα λευκίνη, ισολευκίνη και βαλίνη. Με αυτή την ασθένεια, τα ούρα (και το κερί των αυτιών) έχουν μια γλυκιά μυρωδιά. Επιπλέον, η κλινική εικόνα αυτού του τύπου οργανικής οξυαιμίας περιλαμβάνει υπομελάγχρωση, διακυμάνσεις στην αρτηριακή πίεση, επιληπτικές κρίσεις, έμετο και διάρροια, πτώση των επιπέδων γλυκόζης στο αίμα, κετοξέωση, παραισθήσεις κ.λπ. Το ποσοστό θνησιμότητας στα παιδιά είναι αρκετά υψηλό. σε ενήλικες, χωρίς θεραπεία, μπορεί να συμβεί κώμα και θάνατος λόγω εγκεφαλικού οιδήματος.
Ο αλβινισμός και η αλκαπτονουρία «ενώνονται» μόνο από την τυροσίνη. Ο αλβινισμός, συμπεριλαμβανομένου του οφθαλμοδερματικού, προκαλείται από γενετικές μεταλλάξεις που επηρεάζουν την παραγωγή της χρωστικής μελανίνης. Συγγενείς αλλαγές παρατηρούνται στο γονίδιο TYR στο χρωμόσωμα 11 (11q14.3), το οποίο κωδικοποιεί την τυροσινάση, ένα μελανοσωματικό ένζυμο που περιέχει χαλκό και είναι απαραίτητο για τον σχηματισμό της χρωστικής του δέρματος με βάση τα προϊόντα του μεταβολισμού της τυροσίνης. Αυτή η ασθένεια είναι πολύ πιο συχνή από την αλκαπτονουρία.
Επιπλοκές και συνέπειες
Προκαλούμενη από τη δράση των ενδιάμεσων μεταβολιτών της τυροσίνης - ομογενισικού και βενζοκινονοξικού οξέος - οι συνέπειες και οι επιπλοκές της αλκαπτονουρίας εμφανίζονται λόγω της εναπόθεσης δραστικών χρωματισμένων πολυμερών, της καταστροφής των ινιδίων κολλαγόνου και της επιδείνωσης της κατάστασης του χόνδρου (με μείωση της αντοχής τους στη μηχανική καταπόνηση).
Με την πάροδο των ετών, στην ενήλικη ζωή, αναπτύσσονται εκφυλιστική αρθρίτιδα και οστεοαρθρίτιδα μεγάλων αρθρώσεων (ισχίου, ιερολαγόνιου και γόνατος). Οι μεσοσπονδύλιοι χώροι στενεύουν (ειδικά στην οσφυϊκή και θωρακική μοίρα της σπονδυλικής στήλης) – με ασβεστοποίηση και σχηματισμό οστεοφύτων. Η πυκνότητα του ιστού των υποχόνδριων οστικών πλακών μειώνεται και τα υποκείμενα οστά μπορεί να υποστούν παθολογική αναδιαμόρφωση με σχηματισμό όγκων και παραμορφώσεων. [ 11 ]
Μπορεί να παρατηρηθεί βλάβη στις καρδιακές βαλβίδες (αορτική και μιτροειδής) και στις στεφανιαίες αρτηρίες – με σημάδια στεφανιαίας νόσου, καθώς και σχηματισμός λίθων στα νεφρά και τον προστάτη αδένα – λόγω της ίδιας ασβεστοποίησης. [ 12 ], [ 13 ]
Διαγνωστικά αλκαπτονουρία
Συνήθως, η διάγνωση των συγγενών μεταβολικών διαταραχών βασίζεται στη μελέτη των βιολογικών υγρών του σώματος.
Με βάση ποιες εξετάσεις και αντιδράσεις μπορεί να διαγνωστεί η αλκαπτονουρία; Απαιτούνται εξετάσεις ούρων για την ανίχνευση του ομογεντισινικού οξέος και τον προσδιορισμό του επιπέδου του (φυσιολογικό – 20-30 mg την ημέρα, αυξημένο – 3-8 g). Ένα δείγμα ούρων εξετάζεται με αέρια χρωματογραφία ή φασματομετρία μάζας, χρησιμοποιώντας υγρή χρωματογραφία. Είναι δυνατή μια δοκιμασία διαλογής για την παρουσία χλωριούχου σιδήρου στα ούρα. [ 14 ]
Υπάρχει επίσης μια μέθοδος για ταχεία διάγνωση – ο προσδιορισμός αλκαπτόνης σε αποξηραμένα ούρα σε χαρτί (με βάση την ένταση του χρώματος).
Κατά τη διευκρίνιση της διάγνωσης, η ενόργανη διάγνωση (ακτινογραφία) περιλαμβάνει την αναγνώριση ακτινολογικών σημείων οστεοαρθρίτιδας και άλλων παθολογιών των αρθρώσεων σε ασθενείς.
Η διάγνωση επιβεβαιώνεται με μοριακές γενετικές μεθόδους διάγνωσης κληρονομικών ασθενειών, όπως γενετικές εξετάσεις και αλληλούχιση DNA. [ 15 ]
Διαφορική διάγνωση
Η διαφορική διάγνωση περιλαμβάνει την αιμοχρωμάτωση και την οξεία ηπατική ανεπάρκεια του νεογνού, τη μελανινουρία, την οξεία διαλείπουσα πορφυρία, την αιμοφαγοκυτταρική λεμφοϊστιοκυττάρωση, την πρωτοπαθή μιτοχονδριακή παθολογία, τη ρευματοειδή αρθρίτιδα, την αγκυλοποιητική σπονδυλίτιδα.
Ποιος θα επικοινωνήσει;
Θεραπεία αλκαπτονουρία
Η κύρια θεραπεία για την αλκαπτονουρία είναι η χορήγηση από το στόμα μεγάλων δόσεων (τουλάχιστον 1000 mg την ημέρα) ασκορβικού οξέος. Στα παιδιά, αυτό αυξάνει την απέκκριση του ομογεντισικικού οξέος στα ούρα, και στους ενήλικες μειώνει την περιεκτικότητα στα ούρα του παραγώγου του, του βενζοκινονοξικού οξέος, και επιβραδύνει τη σύνδεσή του με τις δομές του συνδετικού ιστού των αρθρώσεων και το κολλαγόνο. [ 16 ]
Κλινικές της Δυτικής Ευρώπης δοκιμάζουν το φάρμακο Νιτισινόνη (Orfalin), ένα φάρμακο από την ομάδα μεταβολιτών που αναστέλλει το δεύτερο στάδιο του καταβολισμού της τυροσίνης: τον μετασχηματισμό του παρα-υδροξυφαινυλοπυροσταφυλικού σε ομογεντισικό οξύ. Ωστόσο, η χρήση αυτού του φαρμακολογικού παράγοντα οδηγεί στη συσσώρευση τυροσίνης και μπορεί να προκαλέσει σοβαρές παρενέργειες, όπως θολερότητα του κερατοειδούς και φωτοφοβία, ρινορραγίες και αιμορραγία στομάχου, ηπατική ανεπάρκεια, αλλαγές στο αίμα κ.λπ. Παρ' όλα αυτά, στις Ηνωμένες Πολιτείες, η Νιτισινόνη έχει εγκριθεί από τον FDA για τη θεραπεία της τυροσιναιμίας τύπου Ι. [17 ], [ 18 ]
Ως εκ τούτου, η φυσικοθεραπεία – άσκηση για την αύξηση της μυϊκής δύναμης και τη βελτίωση της κινητικότητας των αρθρώσεων, λουτροθεραπεία και πελοειδής θεραπεία για τον περιορισμό του πόνου – εφαρμόζεται για προβλήματα στις αρθρώσεις που προκαλούνται από αλκαπτονουρία.
Παρόλο που η τυροσίνη δεν παρέχεται μόνο από τις τροφές αλλά παράγεται και από τον οργανισμό, συνιστάται στους ασθενείς με αλκαπτονουρία να ακολουθούν μια δίαιτα χαμηλή σε πρωτεΐνες και να περιορίζουν την κατανάλωση τροφών πλούσιων σε τυροσίνη, κυρίως βοδινού και χοιρινού κρέατος, γαλακτοκομικών προϊόντων (ιδιαίτερα τυριών), οσπρίων, ξηρών καρπών και σπόρων.
Πρόληψη
Η πρόληψη των γονιδιακών μεταλλάξεων είναι αδύνατη, αλλά για την πρόληψη της γέννησης παιδιών με υψηλό κίνδυνο συγγενών διαταραχών, υπάρχει ιατρική γενετική συμβουλευτική, η οποία είναι απαραίτητη πριν από μια προγραμματισμένη εγκυμοσύνη για εκείνα τα ζευγάρια των οποίων το οικογενειακό ιστορικό περιλαμβάνει κληρονομικές ασθένειες. [ 19 ]
Πρόβλεψη
Οι θανατηφόρες εκβάσεις από αλκαπτονουρία είναι πολύ σπάνιες και ο θάνατος μπορεί να προκληθεί από σοβαρές επιπλοκές που αφορούν την καρδιά και τα νεφρά. Έτσι, το συνολικό προσδόκιμο ζωής των ατόμων με αλκαπτονουρία είναι καλό.
Αλλά η ποιότητα ζωής μειώνεται λόγω έντονου πόνου στις αρθρώσεις ή τη σπονδυλική στήλη με σημαντικό περιορισμό της κινητικότητας, συχνά προοδευτικό.